Fabrication of functional bioinorganic nanoconstructs by polymer–silica wrapping of individual myoglobin molecules†
Biru
Hu
ab,
Mei
Li
a,
Sajanikumari
Sadasivan
a,
Avinash J.
Patil
a and
Stephen
Mann
*a
aCentre for Organized Matter Chemistry, School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: s.mann@bris.ac.uk
bDepartment of Chemistry and Biology College of Science, National University of Defense Technology, Changsha, 410073, China
First published on 10th December 2010
Abstract
Discrete core–shell hybrid nanoparticles comprising individual met-myoglobin (met-Mb) molecules incarcerated within an ultrathin polymer/silica shell were prepared without loss of biofunctionality by a facile self-assembly procedure. Solubilisation of met-Mb in cyclohexane in the near-absence of water was achieved by wrapping individual protein molecules in the amphiphilic triblock copolymer poly(ethylene-oxide)19–poly(propylene-oxide)69–poly(ethylene-oxide)19 (EO19–PO69–EO19, P123). Addition of tetramethoxysilane to the met-Mb/P123 conjugates in cyclohexane produced discrete nanoparticles that contained protein, polymer and silica, and which were 3–5.5 nm in size, consistent with the entrapment of single molecules of met-Mb. The hybrid nanoconstructs were isolated and re-dispersed in water without loss of secondary structure, and remained functionally active with respect to redox reactions and CO and O2 ligand binding at the porphyrin metallocentre. The incarcerated met-Mb biomolecules showed enhanced thermal stability up to a temperature of around 85 °C. These properties, along with the high biocompatibility of silica and P123, suggest that the silicified protein–polymer constructs could be utilised as functional nanoscale components in bionanotechnology.
1. Introduction
Proteins and enzymes display excellent recognition, binding and catalytic properties that can be exploited to produce a wide range of hybrid functional bionanomaterials1–3 that are of considerable interest in biocatalysis,4 biosensors,5,6 bio-optics,7 biomedical devices8 and drug delivery systems.9–11 However, the fragile nature of many biomolecules seriously constrains their potential application in biotechnology, and as a result, various methods of immobilization and encapsulation have been employed to improve thermal and chemical stabilization without significantly undermining functional properties. For example, proteins have been occluded within mesoporous silicas,12–14 entrapped in sol–gel matrices,15–21 adsorbed onto electrode surfaces,22–26 anchored to organic polymers or multilayers,27–29 and intercalated into layered materials.30–37Whilst the above methods are effective for producing ensembles of protein molecules immobilized in 2- or 3-D inorganic or polymer matrices, they are not readily amenable to the fabrication of discrete protein–inorganic nanoconstructs, which can be dispersed into solvents and used as bio-functionalized sols and fluids. Moreover, isolating single protein molecules within such constructs, for example through the assembly of core–shell bioinorganic nanoparticles, has the potential advantage of minimising deleterious effects on biomolecule function associated with protein–protein interactions and aggregations. Previously we have reported that individual biomolecules such as met-myoglobin (met-Mb), haemoglobin or glucose oxidase38 or single DNA molecules39 can be wrapped with an ultrathin shell comprising condensed clusters of an exfoliated and size-fractionated aminopropyl-functionalized magnesium phyllosilicate organoclay to produce aqueous dispersions of discrete protein–inorganic nanoparticles. Undertaking such procedures in aqueous solutions is constrained by non-specific reactions occurring in the bulk water phase, and this can be circumvented by performing similar processes but within the nanoscale water droplets of reverse microemulsions. For example, single molecules of met-Mb40 or cytochrome c30 have been incarcerated into silica nanoparticles by reaction of tetramethoxysilane (TMOS) at the oil/water interface of surfactant-stabilized microemulsion water droplets. However, only relatively low biomolecule concentrations can be accommodated in the microemulsion droplets due to the onset of mesoscale aggregation41 or macroscopic phase separation, both of which severely undermine the self-assembly of discrete, dispersible protein–inorganic constructs.
Herein we describe a new procedure in which extensive solubilisation of the protein molecules in oil in the near-absence of water is achieved by wrapping individual molecules of met-Mb in the amphiphilic triblock copolymer poly(ethylene-oxide)19–poly(propylene-oxide)69–poly(ethylene-oxide)19 (EO19–PO69–EO19, P123). We then exploit the presence of residual water molecules of hydration associated with the polymer chains to control the hydrolysis and condensation of oil-soluble TMOS molecules specifically at the surface of the met-Mb/P123 nanoconstructs. The resulting silica-entrapped conjugates can be isolated and re-dispersed in water without loss of secondary structure, and significantly, the incarcerated met-Mb biomolecules are thermally stabilized and remain functionally active with respect to redox and gas ligand exchange reactions. As both silica and P123 have relatively high biocompatibility, the silicified protein–polymer constructs could be useful as biocatalysts or biosensors, or as functional components in integrated bionanotechnological devices.
2. Experimental
Sample preparations
0.2 g of the triblock copolymer P123 (EO19–PO69–EO19; Mw = 5760, BASF) was dispersed in 5 g of cyclohexane (Sigma) and stirred for 24 hours to obtain a homogeneous colourless solution. P123/silica nanoparticles were prepared using the above solution by adding 1 g of TEOS (Alfa) to give a P123![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEOS molar ratio of 1:140. The mixture was left stirring for a further 24 hours.
:TEOS molar ratio of 1:140. The mixture was left stirring for a further 24 hours.
Met-Mb was solubilised in cyclohexane as follows. Aqueous solutions of met-Mb (10–12 mg in 15 mL, 0.04 mM) and P123 (37–40 mg in 15 mL, 0.4 mM) were stirred separately for 4 hours and then mixed at 20 °C, stirred for a further 4 hours, and subsequently freeze-dried for 24 hours to produce a brown lyophilized powder containing a Mb:P123 molar ratio of 1:10. The freeze-dried P123–Mb composite (3 mg) was then dispersed in 15 mL of cyclohexane and stirred for 4 hours to give a brown transparent solution.
Preparation of core–shell met-Mb/P123/silica nanoparticle suspensions in cyclohexane was undertaken by adding TMOS (30 µL) to the above Mb/P123 solution and stirring for 24 hours. Typically, the Mb:P123:TMOS molar ratio was 1:10:15. The hybrid nanoparticles were isolated by adding 5 mL methanol to 5 mL of the cyclohexane dispersion to produce a precipitate that was centrifuged, washed with water, and dried at room temperature for 24 hours. The dried powder could be readily resuspended in water. Water with a resistivity of 18.2 MΩ cm was used in all experiments.
Wrapped nanoparticles were also produced by undertaking similar experiments at a constant Mb:TMOS molar ratio but increased P123 concentrations (Mb:P123:TMOS = 1:10:15 to 1:50:15), or at a constant Mb:P123 ratio and increasing TMOS content (Mb:P123:TMOS = 1:10:15 to 1:10:90).
Characterization methods
Samples for transmission electron microscope (TEM) (JEOL 1200EX) and energy dispersive X-ray (EDX) analysis were taken directly from cyclohexane or aqueous solutions and air-dried onto carbon coated copper grids. Samples were unstained except for those prepared from a P123 dispersion in cyclohexane, which were stained with uranyl acetate. Both as-prepared and washed nanoparticles were investigated. UV-Vis spectrometry (Perkin-Elmer Lambda 25) was used to determine Mb concentrations (ε = 1.7 × 105 M−1 cm−1) and record changes in the Mb absorption bands for hybrid nanoparticles dispersed in cyclohexane or water. The thermal stability of native met-Mb in water and met-Mb/P123/SiO2 nanoparticles redispersed in water was determined by temperature-dependent UV-Vis spectroscopy using an attached Peltier temperature control system. Changes in absorption intensity of the 408 nm Soret band of samples allowed to stand for 5 min at specific temperatures between 25 and 85 °C were monitored. Circular dichroism (CD) spectroscopy was used to monitor structural changes in the secondary structure of met-Mb molecules in nanoparticles redispersed in water. CD spectra were recorded at 298 K using a JASCO model 725 spectrometer with a quartz cell and path length of 10 mm at a scan speed of 50 nm min−1. FTIR spectra (Perkin-Elmer Spectrum I) were recorded on washed and air-dried nanoparticles.Thermogravimetric (TG) analysis and differential scanning calorimetry (DSC) (STA409EP) were undertaken on washed, dried samples over a temperature range of 20 to 1000 °C using a heating rate of 5 °C min−1.
Dynamic light scattering (DLS) measurements were performed on a Malvern Zetasizer Nano S equipped with a 4 mW HeNe laser (wavelength = 633 nm) and temperature controller. Measurements were made at 20 °C or 17 °C in water or cyclohexane, respectively, at a backscatter angle of 173° in a low-volume glass cuvette. Refractive indices used were: P123 (1.456, 20 °C), met-Mb (1.45, 20 °C), water (1.33, 20 °C) and cyclohexane (1.4264, 17 °C). The radius of gyration Rg for a collapsed block copolymer chain was estimated from the approximated equation, Rg2 = Nb2/6 where N was the number of repeat units (EO = 38 and PO = 69) and b = 0.6 nm for PEO.42 The model assumed a collapsed architecture based on the internalization of the two co-aligned EO end domains enclosed within a shell of the PO block; i.e., Rg = Rg(EO)19 + [Rg(PO)69]/2.
Functional activity of myoglobin–polymer–silica nanoparticles
Reduction of met-Mb to deoxy-Mb was undertaken by adding 3 mg of sodium dithionite (Na2S2O4, Sigma) to 5 mL of aqueous native met-Mb or aqueous dispersed met-Mb/P123/silica nanoparticles under an argon atmosphere. CO gas was then bubbled through the sodium dithionite-treated solution for less than 5 min at room temperature to produce carboxy-Mb/P123/silica nanoparticles. Subsequent ligand exchange of the bound CO with dioxygen was undertaken by bubbling O2 into the solution for 5 min to produce oxy-Mb/P123/silica nanoconstructs. Binding assays were performed by recording spectral changes in the Soret band using UV-Vis spectrophotometry.3. Results and discussion
TEM images of air-dried, uranyl acetate-stained samples taken from transparent solutions of P123 (EO19–PO69–EO19) in cyclohexane showed ill-defined colloidal aggregates, 40–100 nm in dimension that were composed of discrete electron dense primary particles with an average size of around 2.5 nm (Fig. 1a). Given a P123 molecular weight of 5720 and 107 monomer units, the primary particle size was commensurate with the presence of individual collapsed chains of the triblock copolymer, which had an estimated radius of gyration of 2.1 nm. Addition of TEOS to the above solution at a P123:TEOS molar ratio of 1:140 gave a colourless solution and no evidence of sedimentation within 24 hours. TEM images of unstained samples showed large numbers of discrete electron dense nanoparticles, 19 nm in mean size (Fig. 1b) that contained Si by EDX analysis (Fig. 1b, inset). In contrast, when TEOS was added to cyclohexane in the absence of P123, only a small yield of electron dense particles with a size range between 4.8 and 32 nm (mean = 12.7 nm) was observed, consistent with the general absence of water molecules under these conditions. The above results suggest that P123 molecules can be dispersed in cyclohexane in the form of collapsed molecular chains that act as discrete loci for the formation of silica-entrapped polymer macromolecules. We attribute the specificity of silica nucleation to the presence of residual water molecules associated with the P123 chains, which serve as reaction sites for the controlled hydrolysis and condensation of TEOS molecules. Given these observations, we set out to exploit the above protocol firstly for the solubilisation of native met-Mb in cyclohexane by using P123 as a surface stabilizer, and secondly by employing both P123 and TMOS to wrap individual protein molecules dispersed in the organic solvent with an ultrathin layer of silica.
 | ||
| Fig. 1 (a) Uranyl acetate-stained TEM image of P123 in cyclohexane showing aggregated and discrete polymer nanoparticles. (b) TEM image of P123/TEOS nanoparticles in cyclohexane; inset shows corresponding EDX analysis. (c) TEM image of met-Mb/P123/SiO2 composite nanoparticles in cyclohexane solution. (d) Corresponding size distribution histogram of nanoparticles shown in (c), and (e) corresponding EDX analysis (Cu, Ca and Cr peaks are from sample holder). (f) TEM image of discrete met-Mb/P123/SiO2 nanoparticles re-dispersed in aqueous suspension. | ||
Dispersion into cyclohexane of a lyophilized powder comprising a met-Mb:P123 molar ratio of 1:10 produced a clear brown-coloured solution after stirring for 12–24 hours. Although no electron dense nanoparticles were observed in unstained samples studied by TEM, DLS measurements gave a single peak in the size distribution profile with a hydrodynamic diameter of 3.6 nm, consistent with the presence of molecularly dispersed myoglobin (size = 4.5 × 3.5 × 2.5 nm) (Table S1†).43 In contrast, met-Mb could not be dispersed in cyclohexane in the absence of P123. Addition of TMOS to this solution at a molar ratio of met-Mb:P123:TMOS = 1:10:15 produced no visible changes in optical transparency after 24 hours. TEM images of samples taken directly from the above solution after 24 hours showed discrete electron dense spheroidal nanoparticles (Fig. 1c), with a size range between 3 and 5.5 nm (Fig. 1d), suggesting that individual Mb and P123 molecules were wrapped in an ultra-thin silica sheath. EDX analysis performed on the nanoparticles showed the presence of Fe (6.4 keV), S (2.3 keV) and Si (1.7 keV) (Fig. 1e), confirming the presence of both met-Mb and silica. DLS measurements (Table S1†) gave a hydrodynamic size of 8.5 nm, which increased to 15.6 nm when the met-Mb:P123:TMOS molar ratio was increased from 1:10:15 to 1:10:75, suggesting that protein–polymer constructs with different silica shell thicknesses could be prepared by modifications in the reaction conditions.
The polymer–protein–silica hybrid nanoparticles were extracted from the cyclohexane dispersion by addition of methanol followed by drying to produce a brown powder that could be re-suspended in water as a stable dispersion. TEM images of samples taken from the water suspension showed electron dense nanoparticles with an average size of 4.0 nm (Fig. 1f), and EDX analysis revealed the presence of met-Mb and silica (ESI, Fig. S1a†). The similarity in particle size between samples dispersed in cyclohexane or water was consistent with complete embedding of the protein and polymer molecules within the silica matrix. Changing the met-Mb:P123 ratio from 1:10 to 1:50 at constant TMOS concentration had only a marginal effect on the size of the nanoparticles (ESI, Fig. S1b†). In contrast, the mean size of the hybrid nanoparticles increased from 4.0 to 4.9 nm as the molar ratio of met-Mb:TMOS increased from 1:15 to 1:90 at a constant P123 concentration (ESI, Fig. S1c†). Increasing the TMOS concentration to a met-Mb:TMOS ratio of 1:120 did not produce discrete nanoparticles, instead a network of extraneous silica was observed (ESI, Fig. S1d†).
FTIR spectra of washed and dried met-Mb/P123/SiO2 nanoparticles showed vibration modes at 470 cm−1 (Si–O–Si bend), 1090 cm−1 (Si–O–Si, C–O–C antisymmetric stretch), 1400 cm−1 (C–H bend), 1638 cm−1 and 1617 cm−1 (O–H bending vibration), as well as protein amide I (shoulder at 1655 cm−1) and II (N–H bend/C–N stretch, 1545 cm−1) bands, unequivocally confirming the presence of silica and protein (Fig. 2a and S2a†). The presence of P123 was confirmed by measuring the absorption intensity ratio of the 1090 and 1545 cm−1 bands (A1090/A1545), which increased linearly from 6.29 to 12.5 as the met-Mb:P123:TMOS molar ratios were changed between 1:10:15 to 1:50:15 (Fig. 2b and S2b†). In addition, the A1090/A1545 ratios determined for met-Mb/P123 nanoparticles prepared from the same range of molar ratios but in the absence of TMOS increased from 4.2 to 7.2, confirming that the band at 1090 cm−1 for samples of the met-Mb/P123/SiO2 nanoparticles was associated with the presence of both polymer and silica components (Fig. 2b and S2c†). The results were consistent with TGA profiles of washed and dried met-Mb/P123/silica nanoparticles, which showed mass losses of 3.5%, 52.5% and 2% between 30–100 °C, 100–460 °C, and 460–550 °C, respectively (ESI, Fig. S3†), corresponding in turn to thermodesorption of physically adsorbed water, P123 and Mb decomposition, and residual P123 decomposition. The corresponding DSC trace showed exothermic peaks centered at 167 and 392 °C. FTIR spectra of the residual mass, which was determined from TGA as 40 wt%, showed bands for silica at 474 cm−1 (Si–O–Si bend) and 1095 cm−1 (Si–O–Si antisymmetric stretch) (ESI, Fig. S4†).
![(a) FTIR spectrum of met-Mb/P123/SiO2 nanoparticles; the amide I band at 1655 cm−1 is observed as a shoulder on the strong O–H band at 1638 cm−1. (b) Plot of intensity ratio A1090/A1545 and [P123]/[Mb] molar ratio for Mb/P123 (●) and Mb/P123/SiO2 (■) nanoparticles.](/image/article/2011/NR/c0nr00576b/c0nr00576b-f2.gif) | ||
| Fig. 2 (a) FTIR spectrum of met-Mb/P123/SiO2 nanoparticles; the amide I band at 1655 cm−1 is observed as a shoulder on the strong O–H band at 1638 cm−1. (b) Plot of intensity ratio A1090/A1545 and [P123]/[Mb] molar ratio for Mb/P123 (●) and Mb/P123/SiO2 (■) nanoparticles. | ||
The structural integrity of met-Mb molecules associated with the hybrid nanoparticles was investigated by CD spectroscopy. CD spectra of met-Mb/P123/SiO2 nanoparticles redispersed in water showed bands at 192 and 209 nm, as well as 222 nm, corresponding to amide π–π* and n–π* transitions, respectively, of a α-helical polypeptide secondary structure (Fig. 3). The bands were essentially unchanged in relative intensities compared with aqueous solutions of native met-Mb, indicating that the protein secondary structure remained intact after wrapping with P123 and silica in cyclohexane and transferring to the aqueous phase. UV-Vis spectra of cyclohexane solutions containing met-Mb/P123 or met-Mb/P123/SiO2 nanoparticles showed Soret absorption bands at 413 or 411 nm, respectively, due to π–π* transitions associated with a six-coordinate met-Mb haem group with a strongly bound water molecule44 (Fig. 4(i) and (ii)). The Soret bands were slightly red-shifted from a value of 408 nm recorded for met-Mb in aqueous solution (Fig. 4(iii)). This was consistent with changes in the solvent dipolarity as increasing the thickness of the silica shell resulted in a slight shift of the Soret absorption band back towards 408 nm, and resuspension of the cyclohexane-extracted met-Mb/P123/SiO2 nanoparticles in water gave a band at 409 nm (ESI, Fig. S5†). This interpretation was consistent with UV-Vis spectra of poorly dispersed aggregates of native met-Mb in cyclohexane produced by vigorous stirring, which showed a band at 420 nm associated with changes in solvent polarity (Fig. 4(iv)).
 | ||
| Fig. 3 CD spectra of (i) aqueous met-Mb and (ii) met-Mb/P123/silica hybrid nanoparticles redispersed in water. | ||
 | ||
| Fig. 4 UV-Vis spectra of (i) met-Mb/P123/silica nanoparticles in cyclohexane; (ii) non-silicified Mb/P123 composite in cyclohexane; (iii) aqueous native met-Mb, and (iv) met-Mb suspension in cyclohexane. | ||
To test whether haem functionality in the met-Mb/P123/silica nanoconstructs was retained, we undertook experiments to assess the accessibility of the incarcerated met-Mb molecules to redox agents and gaseous ligands. Addition of a slight stoichiometric excess of solid sodium dithionate to the hybrid nanoparticles in water and under an argon atmosphere resulted in a shift in the Soret band from 409 to 432 nm, indicating reduction of Fe(III) to Fe(II) at the porphyrin centre and formation of deoxy-Mb/P123/silica nanoparticles (Fig. 5). The results confirmed that when placed in water the nanoparticles were porous to small molecules and that the metallocentre remained redox active. Moreover, bubbling CO gas through the suspension of deoxy-Mb/P123/silica nanoparticles produced a blue shift in the 432 peak to a value of 422 nm as well as the appearance of α and β absorption bands at 575 and 536 nm, respectively (Fig. 5), which was consistent with CO binding to the haem group. Subsequently, exposing the wrapped CO-bound deoxy-Mb to O2 resulted in shifts of the 422, 575 and 536 bands to 413, 568 and 530 nm, respectively (Fig. 5, inset), indicating the exchange of CO for O2. The above results were in good agreement with analogous data obtained for native met-Mb in aqueous solution (ESI, Fig. S6†), and indicated that the Mb haem group remained accessible and functional in the Mb/P123/silica composite nanoparticles.
 | ||
| Fig. 5 UV-Vis spectra of met-Mb/P123/silica nanoparticles before (i) and after (ii) adding Na2S2O4. (iii) After CO addition to (ii); (iv) after O2 addition to (iii). Inset: enlarged α and β absorption bands for spectra (iii) and (iv) between 500 and 600 nm. | ||
The thermal stability of aqueous samples of native met-Mb or met-Mb/P123/silica nanoparticles was examined by monitoring changes in the Soret band intensity at 408 nm for temperatures between 25 and 85 °C. Whereas the Soret band intensity for native met-Mb decreased rapidly above 30 °C, only small changes in peak intensity were observed for the met-Mb/P123/silica nanoparticles up to a temperature of 65 °C (Fig. 6 and S7†). Significant degradation was only observed at 85 °C under the conditions employed. The results clearly indicated a significant enhancement in the thermal stability of Mb molecules entrapped within the P123/silica nanoparticles.
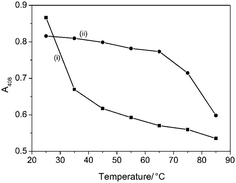 | ||
| Fig. 6 Temperature-dependent plots of absorption intensities of Soret band at A408 for (i) native met-Mb and (ii) met Mb/P123/silica nanoparticles. | ||
4. Conclusions
In conclusion, the preparation of discrete hybrid nanoparticles containing entrapped individual met-Mb molecules has been achieved by controlled silicification of polymer-wrapped protein molecules dispersed in cyclohexane. The method exploits the amphiphilic nature of P123 to solubilize met-Mb in the organic solvent, as well as the presence of residual water molecules of hydration associated with the polymer chains to control the hydrolysis and condensation of TMOS molecules specifically at the surface of the met-Mb/P123 nanoconstructs. The incarcerated protein molecules show retention of their secondary structure and enhanced thermal stability. Moreover, the entrapped met-Mb can be reduced to deoxy-Mb by addition of Na2S2O4, which penetrates the porous silica shell of the hybrid nanoparticles. Similarly, diffusion of gaseous CO or O2 into the nanoparticle interior produces nanoconstructs containing carboxy-Mb and oxy-Mb, respectively. Finally, the use of lyophilized powders of protein/P123 nanocomposites for molecular level dispersion of biomolecules into organic solvents, in association with readily hydrolysable organically functionalized silicon (or titanium) alkoxides may provide a general route to the self-assembly of novel bionanoparticles for use in diverse applications such as drug delivery, biosensing and biocatalysis.Acknowledgements
We thank the EPSRC, UK, and CSC Scholarship Program, China, for funding for M.L. and B.H., respectively, Prof. W. Wu for help with supervising B.H., and Mr A. Brogan for assistance with CD spectroscopy.References
- S. Mann, Nat. Mater., 2009, 8, 781 CrossRef CAS.
- C. A. Mirkin and C. M. Niemeyer, Nanobiotechnology II, Wiley-VCH, Weinheim, 2007 Search PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042 CrossRef CAS.
- U. T. Bornscheuer, Angew. Chem., Int. Ed., 2003, 42, 3336 CrossRef CAS.
- S. A. Yamanaka, B. Dunn, J. S. Valentine and J. I. Zink, J. Am. Chem. Soc., 1995, 117, 9095 CrossRef CAS.
- B. C. Dave, B. Dunn, J. S. Valentine and J. I. Zink, Anal. Chem., 1994, 66, 1120A CrossRef CAS.
- Z. Chen, L. A. Samuelson, J. Akkara, D. L. Kaplan, H. Gao, J. Kumar, K. A. Marx and S. K. Tripathy, Chem. Mater., 1995, 7, 1779 CrossRef CAS.
- E. J. A. Pope, K. Braun and C. M. Peterson, J. Sol-Gel Sci. Technol., 1997, 8, 635 CrossRef CAS.
- D. G. Evans and X. Duan, Chem. Commun., 2006, 485 RSC.
- E. Ruiz-Hitzky, M. Dardar and P. Aranda, J. Mater. Chem., 2005, 15, 3650 RSC.
- A. I. Khan, L. Lei, A. J. Norquist and D. O'Hare, Chem. Commun., 2001, 2342 RSC.
- Y. S. Chaudhary, S. K. Manna, S. Mazumdar and D. Khushalani, Microporous Mesoporous Mater., 2008, 109, 535 CrossRef CAS.
- M. Miyahara, A. Vinu and K. Arig, Mater. Sci. Eng., C, 2007, 7, 232 CrossRef.
- M. Hartmann, Chem. Mater., 2005, 17, 4577 CrossRef CAS.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013 RSC.
- I. Gill, Chem. Mater., 2001, 13, 3404 CrossRef CAS.
- M. L. Ferrer, F. del Monte and D. Levy, Chem. Mater., 2002, 14, 3619 CrossRef CAS.
- I. Gill and A. Ballesteros, Trends Biotechnol., 2000, 18, 282 CrossRef CAS.
- E. H. Lan, B. C. Dave, J. M. Fukuto, B. Dunn and J. Zink, J. Mater. Chem., 1999, 9, 45 RSC.
- D. Avnir, S. Brown, O. Lev and M. Ottolenghi, Chem. Mater., 1994, 6, 1605 CrossRef CAS.
- L. M. Ellerby, C. R. Nishida, F. Nisheda, A. Yamanaka, B. Dunn, J. Valentine and J. Zink, Science, 1992, 255, 1113 CrossRef CAS.
- W. Cao, C. Wei, J. Hu and Q. Li, Electroanal., 2008, 20, 1925 CrossRef CAS.
- Z. Tong, R. Yuan, Y. Chai, Y. Xie and S. Chen, J. Biotechnol., 2007, 128, 567 CrossRef CAS.
- S. Kroning, F. W. Scheller, U. Wollenberger and F. Lisdat, Electroanal., 2004, 16, 253 CrossRef CAS.
- J. Deere, E. Magner, J. G. Wall and B. K. Hodnett, Chem. Commun., 2001, 465–466 RSC.
- E. Topoglidis, A. G. Cass, G. Gilardi, S. Sadeghi, N. Beaumont and J. R. Durrant, Anal. Chem., 1998, 70, 5111 CrossRef CAS.
- A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085 CrossRef CAS.
- V. Panchagnula, C. V. Kumar and J. F. Rusling, J. Am. Chem. Soc., 2002, 124, 12515 CrossRef CAS.
- F. Caruso and C. Schuler, Langmuir, 2000, 16, 9595–9603 CrossRef CAS.
- J. L. Vickery, S. Thachepan, A. J. Patil and S. Mann, Mol. BioSyst., 2009, 5, 744 RSC.
- A. J. Patil and S. Mann, J. Mater. Chem., 2008, 18, 4605 RSC.
- S. C. Holmstrom, A. J. Patil, M. Butler and S. Mann, J. Mater. Chem., 2007, 17, 3894 RSC.
- L. Gao, Q. Gao, Q. Wang, S. Peng and J. Shi, Biomaterials, 2005, 26, 5267 CrossRef CAS.
- A. J. Patil, E. Muthusamy and S. Mann, J. Mater. Chem., 2005, 15, 3838 RSC.
- S. J. Park, T. D. Chung, S. K. Kang, R. A. Jeong, H. K. Boo and H. C. Kim, Anal. Sci., 2004, 20, 1635 CrossRef CAS.
- C. V. Kumar and A. Chaudhari, Chem. Commun., 2002, 2382 RSC.
- C. V. Kumar and A. Chaudhari, J. Am. Chem. Soc., 2000, 122, 830 CrossRef CAS.
- A. J. Patil, E. Muthusary and S. Mann, Angew. Chem., Int. Ed., 2004, 43, 4928 CrossRef CAS.
- A. J. Patil, M. Li, E. Dujardin and S. Mann, Nano Lett., 2007, 7, 2660 CrossRef CAS.
- D. Ma, M. Li, A. J. Patil and S. Mann, Adv. Mater., 2004, 16, 1838 CrossRef CAS.
- E. M. Lambert, C. Viravaidya, M. Li and S. Mann, Angew. Chem., Int. Ed., 2010, 49, 4100 CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, pp. 224–232 Search PubMed.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish and H. Wyckoff, Nature, 1958, 181, 662 CAS.
- M. Ikeda-Saito, H. Horill, L. A. Andersson, R. C. Prince, I. J. Pickering, G. N. George, C. R. Sanders, R. S. Lutz, E. J. McKelvey and R. Mattera, J. Biol. Chem., 1992, 267, 22843.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, UV-Vis, FTIR, DLS data. See DOI: 10.1039/c0nr00576b |
| This journal is © The Royal Society of Chemistry 2011 |