DOI:
10.1039/C0NR00723D
(Paper)
Nanoscale, 2011,
3, 1102-1112
Binary [Cu2O/MWCNT] and ternary [Cu2O/ZnO/MWCNT] nanocomposites: formation, characterization and catalytic performance in partial ethanol oxidation†
Received
28th September 2010
, Accepted 19th November 2010
First published on 23rd December 2010
Abstract
Cuprous oxide agglomerates composed of 4–10 nm Cu2O nanoparticles were deposited on multiwalled carbon nanotubes (MWCNTs) and on ZnO/MWCNTs to give binary [Cu2O/MWCNT] and ternary [Cu2O/ZnO/MWCNT] composites. Di-aqua-bis[2-(methoxyimino)propanoato]copper Cu[O2CCCH3NOMe]2·2H2O 1 in DMF was used as single source precursor for the deposition of nanoscaled Cu2O. The precursor decomposes either in air or under argon to yield CuO2 by in situredox reaction. Thermogravimetric coupled mass spectroscopic analysis (TG-MS) of 1 revealed that methanol formed during the decomposition of 1 acts as a potential in situ reducing agent. Scanning electron microscopy (SEM) of the binary [Cu2O/MWCNT] nano-composite shows an increase of cuprous oxide loading depending on the precursor amount, along the periphery of the MWCNTs as well as formation of larger particle agglomerates. Transmission electron microscopy (TEM) of the sample shows crystalline domains of size 4–10 nm surrounded by an amorphous region within the larger particles. SEM and TEM of ternary [Cu2O/ZnO/MWCNT] clearly reveal that Cu2O nanoparticles are primarily deposited on ZnO rather than on MWCNTs. The catalytic activities of the [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] binary and ternary composites were studied for the selective partial oxidation of ethanol to acetaldehyde with molecular oxygen. While using binary [Cu2O/MWCNT] (13.8 wt% Cu) as catalyst, acetaldehyde was obtained with a yield of 87% at 355 °C (selectivity 96% and conversion 91%). When nanoscale ZnO is present, the resulting [Cu2O/ZnO/MWCNT] composite shows preferential hydrogen and CO2 formation due to the fact that the dehydrogenation and total oxidation pathway is more favoured compared to the binary composite. Significant morphological changes of the catalyst during the catalytic process were observed.
Introduction
Recent studies of carbon nanotubes (CNTs) have focused on tethering metal or metal oxide nanoparticles onto the surface of the nanotubes, forming inorganic hybrids or nanocomposites.1 In such hybrids the catalytic properties are typically not a combination of the individual components but the composite character often results e.g. in higher reactivity and/or selectivity compared to the individual single constituents of the nanocomposite. For example the physical and chemical properties of nanoparticles tethered onto the surface of CNTs can be suitably tuned in accordance with respect to the area of catalytic application by varying the CNT–metal/metal-oxide combination.2 Binary nanocomposites composed of oxides such as TiO2, ZnO, WO3,3Pt/RuO2·H2O, Au, Pt/SiO24 and metals such as Au, Pt and Pd5,6 have thus been synthesized in conjugation with CNTs and used for catalytic and sensoric applications.7
Copper(I) is an especially intriguing example. Cu2O is a low band gap, p-type semiconductor with a direct band gap of 2.12 eV. Its applications are in the fields of photovoltaics, photocatalysis, gas sensors, Li-ion battery research as well as optoelectronic applications.8,9 Cu based catalysts, metallic and oxidic, are used extensively for methanol synthesis from “syngas”,10methanol steam reforming11 and alcohol oxidative dehydrogenation.12 For the latter, metallic copper in association with oxygen is an efficient catalyst. There are two main oxidative reaction pathways: partial oxidation to the corresponding aldehyde (eqn (1)) and complete combustion of the alcohol (eqn (2)):
| | | CnH2n+1OH + 1/2O2 → Cn−1H2n−1CHO + H2O | (1) |
| | | CnH2n+1OH + 3/2nO2 → nCO2 + (n + 1)H2O | (2) |
In studies on the oxidation of methanol and ethanol on Cu (110), it has been shown that the metallic copper surface is especially active when it is in a partially oxidized state.13,14 The ability of metallic copper to adsorb alcohols dissociatively was found to be enhanced by the presence of oxygen (oxygen induced adsorption). The adsorbed nucleophilic oxygen species resulting from dioxygen are able to activate O–H and C–H bonds of ethanol, leading to aldehydes and water formation. However, the oxidation state and the microstructure of the copper catalyst are highly dependent on the gas composition as well as the reaction temperature. Additionally, strong restructuring of the catalyst's surface and the catalyst's bulk structure, even superposed by the redox-behavior of the copper catalyst, emphasizes the complexity of this catalytic system. Thus, a widely accepted view towards the activity of the active copper based catalyst is that in these oxidation reactions its overall activity is a combination of physical and chemical properties, such as oxidation state, surface area, morphology, crystallinity, dispersion and interaction of neighboring active sites, as well as dopant metals on the catalyst. Nearly all of them depend critically on the catalyst preparation techniques employed.15 In addition to the individual synthesis technique used, sintering and restructuring processes of the catalyst material appears, depending upon the time-on-stream in the reactor and the alteration of the copper catalyst dispersion on the support material.16–18 Due to this, catalyst design approaches, such as a mixing in of additives, to hinder catalyst sintering, have been studied extensively.19,20 Recently, CNTs have been suggested as such a dispersion material for catalytic applications.16 However, synthesis of CNT/Cu2O nanocomposites is often hampered by the poor deposition rate of Cu2O particles on the CNT surface, their non-uniform diameters, as well as the poor attachment to the CNT surface which renders their catalytic properties poor. Therefore ways to the synthesis of uniform Cu2O particles tethered onto the surface of MWCNTs are highly desirable.
Herein we report on the synthesis and catalytic performance of binary [Cu2O/MWCNT] and ternary [Cu2O/ZnO/MWCNT] nanocomposites. These composites are comprised of nanoscaled copper oxide as well as zinc oxide which have been generated by a molecular precursor route using the complexes di-aqua-bis[2-(methoxyimino)-propanoato]Cu(II), Cu[O2CCCH3NOMe]2·2H2O 1 and di-aqua-bis[2-(methoxyimino)propanoato]Zn(II) Zn[O2CCCH3−NOMe]2·2H2O 2, which were tethered onto the surface of MWCNTs and subsequently transformed into the binary and ternary title composites. The [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] composites, both contain well defined nanostructured Cu2O oxide sites. By generating nanoscaled Cu2O onto a binary preformed [ZnO/MWCNT] composite, it was possible to study if agglomeration of catalytically active Cu2O particles could be prevented while still maintaining their high dispersion. We could show for the first time that the composite catalysts [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] are active for the partial oxidation of ethanol.
Results and discussion
In the polyol process for the synthesis of Cu2O, the polyalcohol acts as the reducing agent during the formation of Cu2O from a CuII salt. In the case of inorganic copper salts such as copper sulfate, copper chloride and copper nitrate, Cu2O is not formed under hydrothermal conditions as long as copper acetate was not added to the inorganic salts, since the organic acid formed, acts as a reducing agent leading to the formation of Cu2O.23
In contrast, the synthesis of nanoscaled Cu2O can be carried out by using the modified Cu(II) precursor 1 which undergoes a redox decomposition already at moderate temperatures to yield pure Cu2O (Scheme 1). The ligand sphere in 1 facilitates the synthesis of Cu2O without the use of any additional reducing agent. The overall formation of composites [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] therefrom is schematically depicted in Scheme 2.
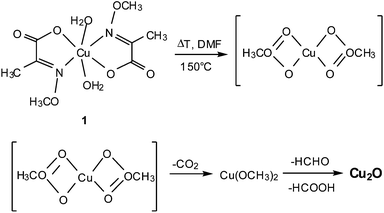 |
| | Scheme 1 Formation route of nanoscaled Cu2O from the oximato complex 1. The isostructural Zn(II) complex 2, decomposes accordingly. | |
![Deposition route to the binary and ternary [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] nanocomposites.](/image/article/2011/NR/c0nr00723d/c0nr00723d-s2.gif) |
| | Scheme 2 Deposition route to the binary and ternary [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] nanocomposites. | |
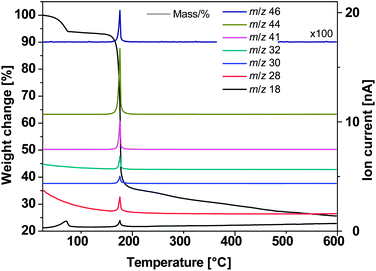 |
| | Fig. 1
TG-MS of Cu–oximato complex 1. | |
Based on the solid product formed and the fragments detected by mass spectrometry, the thermal decomposition reaction for the formation of Cu2O can be formulated as shown in Scheme 1. The product, copper carboxylate has been proposed based on the decomposition of the similar Zn oximato complex.21
The Cu2O obtained from a solution phase decomposition in DMF was analysed for the residual C, H and N content, with values found to be 7.78%, 0.73%, 5.50%, respectively. A possible reason for the observed residual C, H, N content might be due to the adsorbed acetonitrile produced during the synthesis step (Fig. 2). The complete decomposition of 1 takes place only above 186 °C and during the present synthesis step a temperature of 153 °C was maintained which further accounts for the observed organic residue being adsorbed on the surface of the solid product. The organic residue might act as an additional reducing agent in further conversion of CuI to Cu0 with increasing temperature (see ESI†, Fig. 1).
![IR spectra of (a) MWCNT, (b) Cu2O and (c) binary [Cu2O/MWCNT] nanocomposite.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f2.gif) |
| | Fig. 2
IR spectra of (a) MWCNT, (b) Cu2O and (c) binary [Cu2O/MWCNT] nanocomposite. | |
Thermogravimetry and BET surface area of [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] composites
The binary and ternary composites [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] are studied for their stability in air. Their stability decrease by ∼135 °C for lower Cu2O loading and ∼250 °C for higher loading compared to bare MWCNTs (see Experimental section and ESI†, Fig. 2).
Importantly, the composites however, are stable at the typical operating temperature of the copper catalysts in partial oxidation reactions. Table 1 summarizes copper loading, particle size of Cu2O, decomposition temperature and surface area of the as synthesized composites.
Table 1
Copper content, Cu2O particle size, decomposition temperature and surface area of MWCNT, Cu2O, [Cu2O/MWCNT] and ternary [Cu2O/ZnO/MWCNT] composites as catalysts
| Samples |
Cu content from synthesis/wt% |
Decomposition temperature/°C |
Cu2O size SEM dCu2O/nm |
BET surface area/m2 g−1 |
| MWCNT |
— |
642 |
— |
16 |
|
[
Cu
2
O
/MWCNT]
|
3.09 |
506 |
30 |
— |
| 6.00 |
– |
52 |
— |
| 8.74 |
503 |
57 |
— |
| 11.32 |
– |
56 |
— |
| 13.76 |
387 |
73 |
22 |
|
[
Cu
2
O
/
ZnO
/MWCNT]
|
13.76 |
385 |
8 |
— |
| 10 (ZnO) |
|
Cu2O
|
— |
— |
132 |
24 |
The BET surface area of bare MWCNTs, unsupported Cu2O, and the [Cu2O/MWCNT] composite (19.5 mmol loading based on 1) are determined to be 16 m2 g−1, 24 m2 g−1 and 22 m2 g−1, respectively, by physisorption measurements with nitrogen. The slight increase of surface area in the sample deposited with Cu2O in comparison with bare MWCNT reveals additional active surface area due to the presence of the copper oxide on the MWCNT. A similar increase in surface area has been found in our previous study with Au and ZnO deposited MWCNT materials.7a Both catalyst systems generate acetaldehyde as major product. However, the significant higher H2 selectivity indicates that the [Cu2O/ZnO/MWCNT] ternary system favors the dehydrogenation to a higher content than the binary [Cu2O/MWCNT] composite system.
Infrared spectroscopy (IR) of [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] composites
The IR absorptions at around 628 cm−1 can be attributed to the lattice vibration of the Cu2O (Fig. 2).24 During the decomposition of 1acetonitrile is produced and can coordinate to the surface of the Cu2O leading to an absorption peak corresponding to nitrile at 2150 cm−1. The IR spectra of [Cu2O/ZnO/MWCNT] (not shown here) show similar characteristics to those of binary [Cu2O/MWCNT].
Structural characteristics of both composites, as well as the individual components of both composites have been investigated by X-ray powder diffraction (XRD) (Fig. 3a–d). Fig. 3a shows the diffractogram for bare MWCNT (Mo-Kα) indicating some amorphous carbon besides the typical graphitic pattern for CNTs [ICDD PDF card no. 25-284, see also ESI†, Fig. 3a for individual MWCNT spectrum]. The composite materials derived therefrom show all the XRD peaks for the constituents of the composites. The broad signals for the Cu2O (Co-Kα) [ICDD PDF card no. 74-1230] in the bare Cu2O sample indicate its crystalline, nanoparticulate state (b). From the TEM analysis of Cu2O it is indicative that the individual particles consisting of small crystallites are individually surrounded by an amorphous material (see Fig.8). In the case of the ternary [Cu2O/ZnO/MWCNT] composite, mainly ZnO [ICDD PDF card no. 36-1451] reflexes were observed (Fig. 3d). This is probably due to the higher amount of the ZnO on the surface of the MWCNTs. However, the diffraction peak at 2θ, 49.76°, corresponds to the diffraction of the [200] plane of Cu2O, thereby confirming the additional presence of Cu2O in the ternary composite (see also ESI†, Fig. 3 for individual spectrum).
![X-Ray powder diffraction pattern of (a) MWCNTs (Mo-Kα), (b) Cu2O derived from 1 (Co-Kα), (c) [Cu2O/MWCNT] and (d) [Cu2O/ZnO/MWCNT] nanocomposite catalysts, (°) ZnO and (#) Cu2O.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f3.gif) |
| | Fig. 3
X-Ray powder diffraction pattern of (a) MWCNTs (Mo-Kα), (b) Cu2O derived from 1 (Co-Kα), (c) [Cu2O/MWCNT] and (d) [Cu2O/ZnO/MWCNT] nanocomposite catalysts, (°) ZnO and (#) Cu2O. | |
![X-Ray photoelectron spectra for Cu 2p1/2 and Zn 2p3/2 and Zn 2p1/2 and Zn 2p3/2 in binary [Cu2O/MWCNT] (solid line) and ternary [Cu2O/ZnO/MWCNT] (dashed line).](/image/article/2011/NR/c0nr00723d/c0nr00723d-f4.gif) |
| | Fig. 4
X-Ray photoelectron spectra for Cu 2p1/2 and Zn 2p3/2 and Zn 2p1/2 and Zn 2p3/2 in binary [Cu2O/MWCNT] (solid line) and ternary [Cu2O/ZnO/MWCNT] (dashed line). | |
The O 1s photoelectron spectra of the [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] composites are shown in Fig. 5 respectively. The C 1s spectrum of bare MWCNTs shows the graphitic carbon peak at 284.5 eV as reported previously for MWCNTs.7 The O 1s signals of [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] show different profiles. The binding energy of O 1s for both composites is observed at 530.9 ± 0.3 eV which can be attributed to the oxygen associated with Cu and Zn oxide.26 The shoulder around ∼532.7 eV of BE corresponds to organic functionalities (e.g. C–OH groups) associated with MWCNTs27 and are due to their original purification process (e.g. acid treatment to remove catalyst particle).
SEM images of Cu2O powder and of [Cu2O/MWCNT] composite with different loading of Cu2O nanoparticles are shown in Fig. 6a–f. The Cu2O powder (Fig. 6a) shows irregular to spherical particles with an average size of 132 nm. Fig. 6b–f shows Cu2O particles on MWCNTs at different amounts of CuO2 loadings. It was found that the Cu2O loading has a linear relationship with the amount of precursor complex 1 used. The particle size increases from 30–72 nm from the lowest to the highest amount of precursor, but for the intermediate amounts of 1 the particle size remains nearly constant (∼50 nm). There is also evidence from the loading behaviour that particle growth takes place as the precursor amount increases due to Ostwald ripening, instead of an increase in the peripheral distribution of the deposited, similar sized Cu2O particles alongside the MWCNTs. The cuprous oxide particles tend to deviate from the spherical size and grow in the form of aggregate lumps directly on the surface of the CNTs. It can also be seen that in some cases the tip of the CNT is also decorated (Fig. 6c). This is also confirmed by TEM (see next section).
![SEM image of (a) Cu2O powder derived from 1 and of Cu2O tethered to MWCNTs in [Cu2O/MWCNT] at different loadings of 1: (b) 3.9, (c) 7.8, (d) 11. 7, (e) 15.6 and (f) 19.5 mmol. (Scale bar: 300 nm.)](/image/article/2011/NR/c0nr00723d/c0nr00723d-f6.gif) |
| | Fig. 6
SEM image of (a) Cu2O powder derived from 1 and of Cu2O tethered to MWCNTs in [Cu2O/MWCNT] at different loadings of 1: (b) 3.9, (c) 7.8, (d) 11. 7, (e) 15.6 and (f) 19.5 mmol. (Scale bar: 300 nm.) | |
A SEM image of Cu2O deposited on a binary [ZnO/MWCNT] is shown in Fig. 7a and b. Therefrom the observation is that Cu2O is mainly deposited on ZnO in the binary [ZnO/MWCNT] composite. This is due to the fact that the surface of the MWCNTs is not uniformly functionalized and hence is not available for tethering of nanoparticles. Thus, Cu2O will preferentially deposit on the previously formed ZnO nucleation sites. In the present case ZnO acts as such a nucleation site for the formation of Cu2O.
![(a) and (b) SEM images of [Cu2O/MWCNT] at two different magnifications.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f7.gif) |
| | Fig. 7 (a) and (b) SEM images of [Cu2O/MWCNT] at two different magnifications. | |
 |
| | Fig. 8 (a) TEM and (b) HRTEM of the Cu2O powder obtained from thermal decomposition of the Cu–oximato precursor 1. Inset in (b) shows the SAED pattern of the nanocrystalline Cu2O powder. | |
Decomposition of the precursor 1 in the presence of MWCNTs results in uniform decoration of Cu2O on the surface of the MWCNTs (Fig. 9a). Cu2O agglomerates of size 30–70 nm consisting of 4–10 nm crystalline domains of Cu2O were deposited. These are evenly distributed alongside the periphery of the MWCNTs (Fig. 9a) as well as decorated on the tip of the tubes (Fig. 9b). The Cu2O aggregates seem to contain crystalline domains which are embedded inside an amorphous material (Fig. 9b, inset) similar to the Cu2O powder shown in the Fig. 8.
![TEM and HRTEM of the [Cu2O/MWCNT] nanocomposite obtained from the thermal decomposition of 1. (a) [Cu2O/MWCNT] composite overview (b) indicating the tip coverage of the CNT. Inset in (b) HRTEM of a Cu2O nanoparticles on MWCNT. (Scale bar 5 nm.)](/image/article/2011/NR/c0nr00723d/c0nr00723d-f9.gif) |
| | Fig. 9
TEM and HRTEM of the [Cu2O/MWCNT] nanocomposite obtained from the thermal decomposition of 1. (a) [Cu2O/MWCNT] composite overview (b) indicating the tip coverage of the CNT. Inset in (b) HRTEM of a Cu2O nanoparticles on MWCNT. (Scale bar 5 nm.) | |
TEM micrographs of the [Cu2O/ZnO/MWCNT] nanocomposite are shown in Fig. 10a and b. It is observed that the Cu2O particles were mainly formed on the previously deposited ZnO (Fig. 10a and inset) as observed in the case of SEM images. The so formed Cu2O is irregular in shape and covers the surface of the ZnO. It is also interesting to note that the ZnO previously deposited is not affected (in shape and size) during the thermal decomposition process and is tethered to the MWCNTs. ZnO as previously reported by our group has a narrow size below 10 nm.7b The morphology of the deposited Cu2O is appearing to be of flaky type, sitting on the ZnO particles which are present on the MWCNTs compared to MWCNT alone.
![TEM and HRTEM of the [Cu2O/ZnO/MWCNT] nanocomposite obtained from consecutive thermal decomposition of Zn and Cu oximato complexes. (a) [Cu2O/ZnO/MWCNT] composite overview (inset as well) (b) oval shaped ZnO particle which has been deposited and on which irregularly shaped Cu2O is deposited consecutively.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f10.gif) |
| | Fig. 10
TEM and HRTEM of the [Cu2O/ZnO/MWCNT] nanocomposite obtained from consecutive thermal decomposition of Zn and Cu oximato complexes. (a) [Cu2O/ZnO/MWCNT] composite overview (inset as well) (b) oval shaped ZnO particle which has been deposited and on which irregularly shaped Cu2O is deposited consecutively. | |
Catalytic activity of binary [Cu2O/MWCNT] and ternary [Cu2O/ZnO/MWCNT] composites in the partial oxidation of ethanol
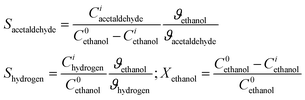
The [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] nanocomposites were tested towards their catalytic activity and selectivity with regard to the partial oxidation of ethanol. Table 2 gives an overview of the catalytic properties for the different composite catalysts. The catalyst activity (ethanol conversion) and selectivity (acetaldehyde formation) are also indicated in Table 2. The given values are related to the third temperature programmed (TP) reaction cycle.
Table 2 Results of ethanol oxidation with [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] composites as catalysts. mcat = 10 mg; T = 140–500 °C; Vtot = 100 ml min−1; 6 vol% ethanol and 3 vol% oxygen. Used abbrevations: X = conversion, S = selectivity, Y = yield, ci = concentration of the respective component, ϑ = stoichiometric coefficienta
| Samples |
Cu content/wt% |
Cu2O size dCu2O/nm |
X ethanol (%) |
S acetaldehyde (%) |
Y acetaldehyde (%) |
Y
hydrogen (%) |
 . .
|
| MWCNT |
— |
— |
65 (524 °C) |
82 (524 °C) |
53 (524 °C) |
— |
|
[
Cu
2
O
/MWCNT]
|
3.1 |
30 |
89 (400 °C) |
93 (400 °C) |
83 (400 °C) |
8 (507 °C) |
| 8.7 |
57 |
91 (360 °C) |
87 (360 °C) |
79 (360 °C) |
7 (507 °C) |
| 13.8 |
73 |
91 (355 °C) |
96 (355 °C) |
87 (355 °C) |
7 (491 °C) |
|
[
Cu
2
O
/
ZnO
/MWCNT]
|
13.8 |
8 Cu2O |
95 (375 °C) |
93 (375 °C) |
88 (375 °C) |
39 (507 °C) |
| |
10 ZnO |
|
|
|
|
Table 2 shows that MWCNTs alone are already active for the oxidative dehydrogenation of ethanol (for a graphical sketch, see ESI†, Fig. 4). Furthermore, it can be observed that the [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT] nanocomposites show high activities and selectivity for the partial oxidation of ethanol. At 375 °C the yield was 88% of the desired product, acetaldehyde.
The variation of the Cu content in the [Cu2O/MWCNT] composite and the Cu particle size shows no significant trend concerning the catalyst performance.
However, with an increase in Cu content, the temperature at which ethanol conversion reaches a maximum, shifts to lower values. This again implies that a higher Cu content leads to higher activity (Table 2) (note: a structure sensitive reaction can be ruled out, since the reaction does not change markedly with catalyst particle size, see Table 2, the maximum of the selectivity/conversion efficiency shifts to lower values with increasing Cu content and is not affected by the copper particle size). The comparison between the [Cu2O/MWCNT] (Cu 13.8 wt%) and the [Cu2O/ZnO/MWCNT]catalyst shows little difference with regard to the catalyst performance. Using the ZnO modified nanocomposites it can be seen that the conversion of ethanol increases to 95% whereas the selectivity decreases slightly to 93%. Note, that the maximum of the catalyst activity is shifted to higher temperatures in the case of the Cu/ZnO system. Although the differences are small as said, their reproducibility was very good (relative deviation <5%).
Taking a closer look at the volume fractions of the products during reaction (see Fig. 11) it can be observed that the volume fraction of hydrogen and CO2 is significant higher for the Cu/ZnO catalyst system above 400 °C, resulting in a lower selectivity. This indicates that the Cu/ZnO-system favours both dehydrogenation and the total oxidation pathway compared to the [Cu2O/MWCNT]catalyst. Fig. 12 shows the selectivity and conversion rates of the binary and ternary composites with respect to acetaldehyde.
![Volume fraction as a function of temperature for the third cycle of the TP-reaction of ethanol on 10 mg of pre-reduced [Cu2O/MWCNT] (top) and [Cu2O/ZnO/MWCNT] (bottom). The molar ratio of ethanol to oxygen is 1 : 1.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f11.gif) |
| | Fig. 11 Volume fraction as a function of temperature for the third cycle of the TP-reaction of ethanol on 10 mg of pre-reduced [Cu2O/MWCNT] (top) and [Cu2O/ZnO/MWCNT] (bottom). The molar ratio of ethanol to oxygen is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. :1. | |
![Selectivity S of acetaldehyde, conversion percentage X of ethanol and yield Y of acetaldehyde for the TP-reaction of ethanol each on 10 mg of pre-reduced [Cu2O/MWCNT] (top) and [Cu2O/ZnO/MWCNT] (bottom).](/image/article/2011/NR/c0nr00723d/c0nr00723d-f12.gif) |
| | Fig. 12 Selectivity S of acetaldehyde, conversion percentage X of ethanol and yield Y of acetaldehyde for the TP-reaction of ethanol each on 10 mg of pre-reduced [Cu2O/MWCNT] (top) and [Cu2O/ZnO/MWCNT] (bottom). | |
Remarkably, in all these TP-reaction experiments, at certain temperatures an abrupt increase in the catalyst activity is observed. At first the conversion of ethanol increases slightly but at approximately 300 °C [Cu2O/MWCNT] and at 325 °C for [Cu/ZnO/MWCNT], a stepwise enhancement of the catalyst activity is observed. From the methanol oxidation it is well known that the enhanced activity of the Cu catalyst coincidences with the transformation of the oxidic phase to the suboxidic/metallic phase observed by the O K- and Cu L3-NEXAFS spectra.12 Thus, it can be assumed that the observed enhanced activity of the used Cu composites indicates probably a similar phase change for the binary [Cu2O/MWCNT] and ternary [Cu/ZnO/MWCNT]catalysts.
In order to determine the phase change occurring during the TP-reaction, the [Cu2O/MWCNT] composite catalyst was taken off after each processing stage. First after H2reduction (6 vol% H2, 400 °C, t = 30 min), secondly after H2reduction and following ethanol treatment (6 vol% EtOH, 140–500 °C, β = 10 K min−1) without oxygen and thirdly after all three catalysis cycles. After each individual procedure the catalysts were analysed by XRD (Fig. 13a–d). As expected, during the initial H2reduction step complete reduction of Cu2O takes place and leads to metallic elemental copper (Fig. 13b). Only the metallic copper phase is present on the MWCNTs. In the second stage, when H2reduction had taken place, and after ethanol treatment without the presence of oxygen, no phase change of the metallic copper obtained in the previous reduction step was observed (Fig. 13c). This indicates that ethanol vapour alone had no effect on the already existing metallic copper surface or the Cu0 species formed. During the ethanol treatment, without oxygen in the reactor feed, no conversion of the ethanol is observable. This proves that the Cu0 species alone, without the presence of oxygen is not an active catalyst for the dehydrogenation of ethanol. However, in order to obtain acetaldehyde with a Cu-catalyst the presence of oxygen is necessary. The same catalyst analysed after the three TP-reaction cycles shows the presence of Cu2O in addition to metallic copper (Fig. 13d). During reaction, the metallic copper phase and Cu0 species accordingly react with the oxygen in the reaction gas mixture whereas under the chosen conditions only Cu2O and not CuO was observable. Under these conditions both Cu0- and CuI-species are co-existing. For the methanol oxidation it has indeed been shown on elemental copper that Cu2O catalyses the non-selective total oxidation of methanol.28 Furthermore, after three reaction cycles the XRD signals, corresponding to graphitic carbon were seen indicating that carbon nanotubes are stable at these operating conditions. The active phase of the catalyst is supposed to be a metastable Cu-suboxide and is thus only detectable with in situ techniques.12 It should be noted that with the ex situXRD analysis of the used catalyst it was not possible to distinguish the phase change of the catalyst, correlating with an abrupt activity enhancement.
![X-Ray powder diffraction pattern (Co-Kα) of [Cu2O/MWCNT] (a) as synthesized, (b) after H2reduction (c) after H2reduction followed by ethanol treatment and (d) after three cycles of oxidative dehydrogenation reaction (*) Cu, (#) Cu2O and (+) MWCNT.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f13.gif) |
| | Fig. 13
X-Ray powder diffraction pattern (Co-Kα) of [Cu2O/MWCNT] (a) as synthesized, (b) after H2reduction (c) after H2reduction followed by ethanol treatment and (d) after three cycles of oxidative dehydrogenation reaction (*) Cu, (#) Cu2O and (+) MWCNT. | |
The morphological changes occurring during the same stages of the TP-reaction as mentioned above were studied by SEM and TEM (Fig. 14a–d) The initial H2reduction does not induce any particle growth and the shape of the copper formed remains more or less as that of the previously deposited Cu2O (Fig. 14a), implying the copper particles are tethered to the carbon nanotube surface without any further movement along the CNT surface of under this condition. The particle size is about 20 nm (determined by TEM as well from the XRD by the Scherrer equation). After the catalytic reaction, significant particle growth had taken place. The process of agglomeration continues with each catalytic cycle and at the end of three cycles bigger particles as in Fig. 14c and d are observed. It has been observed for copper on graphitic fibres that, depending on where the metal is deposited (at the edges or on the basal plane), it is possible that a particle morphology change can take place. It also proposed that in a particular gas environment, interaction of support medium can influence metal particle morphology.29 Thus in the present case, the copper resulting from Cu2O in the H2 environment remains similar in morphology on the carbon nanotube as that of the synthesized material. When ethanol is passed, a particle rounding takes place, indicating the subtle dependence of the catalyst particle morphology on the reaction conditions. In addition, there exists a direct relationship between the mobility temperature of small particles of ∼10 nm in diameter on the graphitic basal plane and bulk melting point of the metal.29 Thus, during ethanol treatment as well as during catalytic cycles, a surface atom movement of copper might occur on the CNT surface, leading to particle growth. Due to a weak {Cu-basal plane/CNT} interaction,29 bigger catalyst particles which form via agglomeration, can even detach from the surface of the CNTs. TEM analysis of a catalyst particle after three TP-reactions is shown in Fig. 15a and b. The presence of an amorphous layer with a thickness of approximately 5 nm around a dense copper core is observed and remarkable. Here, the existence of metastable Cu-suboxide can be ruled out, due to its instability and further conversion to the stable Cu2O. Moreover, if formed at all, Cu2O would be recognizable in the micrograph due to its stronger contrast in TEM compared to carbon. It is noteworthy that there is no contrast difference of the amorphous shell on the particle with respect to the CNT support material on which it is still tethered. Thus the catalytic working state can be considered to be elemental copper with an amorphous layer. It was found by EXAFS that copper is in a strained nanocrystalline form of the metal, while active.30 However, the metal does not consist of large crystallites and contains a defect structure in which oxygen is already intercalated.
![SEM images of [Cu2O/MWCNT] composite (prepared from 19.5 mmol of 1) (a) after H2reduction only, (b) after H2reduction followed by ethanol treatment, (c) after catalysis and (d) after catalysis showing regions of bigger particles (scale bar: 800 nm).](/image/article/2011/NR/c0nr00723d/c0nr00723d-f14.gif) |
| | Fig. 14
SEM images of [Cu2O/MWCNT] composite (prepared from 19.5 mmol of 1) (a) after H2reduction only, (b) after H2reduction followed by ethanol treatment, (c) after catalysis and (d) after catalysis showing regions of bigger particles (scale bar: 800 nm). | |
![TEM of the [Cu2O/MWCNT]catalyst after reaction (a) overview of the agglomerated copper particle and (b) higher magnification of the outer surface of the particle.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f15.gif) |
| | Fig. 15
TEM of the [Cu2O/MWCNT]catalyst after reaction (a) overview of the agglomerated copper particle and (b) higher magnification of the outer surface of the particle. | |
For a binary Cu/ZnO system, its activity and hydrogen selectivity has been correlated with Cu–Zn interactions.31 Activity of the catalyst and yield of hydrogen increased with copper content for low loadings, but then decreased, when copper loading was further increased. However, an optimum of the Cu/Zn ratio might be responsible for both activity and hydrogen yield. Using the [Cu2O/ZnO/MWCNT] ternary composite in the partial oxidation of ethanol, significant more hydrogen was detected. This indicates, introducing additional ZnO into the binary [Cu2O/MWCNT] composite, the reaction pathway is altered, thereby changing the resulting final product.
Conclusions
We have demonstrated synthesis, characterization and catalytic oxidative dehydrogenation of ethanol using binary and ternary nano-composite catalysts of the type [Cu2O/MWCNT] and [Cu2O/ZnO/MWCNT]. Cu2O and ZnO have been deposited using a single source molecular type precursor by thermal decomposition. The control on copper as well as on Cu2O loading can be achieved by varying the precursor amount, which had a slight influence on the particle size of the Cu2O formed on the MWCNTs. The Cu content had no influence on the activity and the selectivity in the case of [Cu2O/MWCNT] composite. The conversion of Cu2O to metallic copper takes place during the initial H2reduction step, which remains in the native state during ethanol treatment. After reaction both Cu0 and CuI are present in the catalyst. During catalysis the particle size increases and densification occurs. Thus the [Cu2O/MWCNT] composite provides a valuable source for the preparation of a highly disperse and active catalyst for dehydrogenation of ethanol.
Experimental section
General
Synthesis of precursor 1 and MWCNT based composites were carried out under air if not specified otherwise. Solvents were used as received and Milli-Q water was used in all the cases of solution preparation. Copper acetate monohydrate (Fluka 99%), pyruvic acid [CH3COCOOH] (Fluka 98%), O-methylhydroxylamine hydrochloride [CH3ONH2·HCl] (Merck KGaA, 98%), sodium bicarbonate (Fluka, analytical grade), multiwalled carbon nanotubes (Electrovac Klosterneuberg, Austria) were used as received. Dimethylformamide (DMF, Merck KGaA) was degassed before use. Synthesis of the binary [ZnO/MWCNT] composite used for the synthesis of [Cu2O/ZnO/MWCNT] was described in detail.7b
Synthesis of di-aqua-bis[2-(methoxyimino)propanoato]2copper 1
Synthesis of Cu oximato complex has been carried out in similar lines as described for Zn–oximato complex.21 A sample of 5.0 g of copper acetate dihydrate (25 mmol) in 25 ml water was added to an in situ prepared oxime ligand in water. The ligand was prepared by taking 50 mmol each of pyruvic acid (4.40 g, 50 mmol) and O-methylhydroxylamine hydrochloride (4.18 g, 50 mmol) according to the procedure described by Spencer et al.22 The reaction mixture was stirred for further 4 h, during which a pale green product was formed. The product was filtered and washed with water and dried at 50 °C to give pure substance. Yield 2.26 g (45.3%) MP ≥ 182 °C (decomp). EI-MS: m/z (relative abundance in %): CH3OH 32 (5), CH3CN, 41 (45), CO2 44 (100), Cu 63 (5), 72 (5), 104 (20), 117 (5), 145 (20). CHN combustion analysis: C8H16N2O8Cu (%): calc. N: 8.44, C: 28.94, H: 4.82; found N: 7.95, C: 27.7, H: 4.7.
Synthesis of [Cu2O/MWCNT], [Cu2O/ZnO/MWCNT] composites and Cu2O powder
30 mg MWCNTs (2.5 mmol) were dispersed in 20 ml of DMF and sonicated for 15 min. A defined amount (5–25 mg, 15–75 µmol) of 1 was added and the mixture was stirred until the complex dissolved (maximum solubility of copper oximato complex in DMF is 0.33% (w/v)). The resulting mixture was refluxed for 3 h yielding a dispersion of cuprous oxide deposited MWCNT (binary [Cu2O/MWCNT] composite). The product was filtered, washed with ethanol and dried at 50 °C for several hours. The synthesis can be scaled up to higher quantities (several grams). [Cu2O/ZnO/MWCNT] composites were synthesized by consecutive deposition of ZnO and Cu2O from Zn–oximato and Cu–oximato precursors from DMF solution. In a typical experiment 30 mg of [ZnO/MWCNT] was taken in DMF (20 ml) stirred for several minutes for good dispersion. 15–25 mg of 1 was added and completely dissolved. The mixture is processed in a similar way as described above to give ternary composite [Cu2O/MWCNT]. Cu2O powder was synthesized, typically, in large scale by taking for e.g. 1.5 g of 1 in 300 ml of DMF and refluxing for above mentioned time, during which a reddish yellow powder was obtained. The product was processed similarly as that of the composites.
Sample characterization
IR spectroscopic measurements were carried out using a Nicolet FTIR spectrometer. Thermogravimetric coupled mass spectrometric (TG-MS) studies were performed using the instrument from Netsch. 10–12 mg of the precursor complex was heated at a heating rate of 10 K min−1 in helium atmosphere. The stability of the [Cu2O/MWCNT] composite was analyzed by taking 6.0 ± 0.5 mg of different Cu2O loaded material and heating in air. BET surface area measurements were carried out with Nova 3000e surface area and pore size analyzed by nitrogen sorption method. X-Ray powder diffraction (XRD) of the composites was performed using StadiP instrument from StadiP, Stoe & Cie GmbH, Darmstadt in Debye–Scherrer mode (flat specimen, transmission mode) with Co-Kα1-radiation with a Ge(111) monochromator. The diffractograms were compared with the reported ICDD database. XPS measurements were performed using ESCA lab 250 (thermo VG scientific) with monochromated Al-Kα-radiation in constant analyzer energy mode (CAE) with a pass energy of 50 eV for all spectra. SEM investigations were performed with Philips XL 30 FEG microscope at 20 keV. HRTEM investigations were carried out using CM20FEG (Philips) at 200 kV to determine the size and morphology of the Cu2O particle. Samples were prepared in high purity ethanol and dripped on lacy-carbon copper grid.
Catalytic measurements apparatus
The apparatus consists of three main parts: the gas dosing unit, the reactor and the online analytics. The gas dosing took place with mass-flow controllers (Bronkhorst) and two-stage gas saturators enabled the enrichment of liquid components e.g.ethanol in the carrier gas flow. The partial oxidation of ethanol was performed in a tubular reactor (inner diameter 4 mm) consisting of quartz glass in which the catalyst (10 mg) was fixed with a quartz wool plug. The reaction temperature was controlled by an electrically heated oven and the sample thermo-couple was placed in the catalyst bed to measure the reaction temperature. Online analysis of the gaseous components at the reactor outlet was performed by a quadrupole mass spectrometer (InProcess Instruments, GAM 400). Before the actual measurement took place an extensive calibration with each component was carried out. The calibration itself was based on a complex matrix involving the dependencies between the ion currents and the gas concentrations as well as the fact that some components have peaks at the same mass number. All this is implemented in a software routine. However, the application of such a calibration matrix is well known.
Temperature programmed (TP) reaction
All experiments were performed under continuous flow (overall volume flow 100 ml min−1, standard conditions) and at ambient pressure. The reaction gas mixtures were balanced with argon to give a final flow of 100 ml min−1. As model reaction the partial oxidation of ethanol to acetaldehyde was investigated to determine the catalyst activity. In order to obtain the start-up phase of the catalyst, prior to the temperature programmed reaction (TP-reaction) a reduction with 6 vol% H2 at 400 °C (30 min) has been carried out. After the isothermal reduction the temperature was cooled to 140 °C and TP-react experiments were carried out. The volume fraction of ethanol (6 vol%) and the volume fraction of oxygen (3 vol%) at the reactor inlet was held constant whereas the catalyst was heated to 500 °C with a heating rate of 10 K min−1. This procedure was performed overall three times (see Fig. 16) to achieve steady state and to stabilize the catalytic system (activity and selectivity to acetaldehyde).
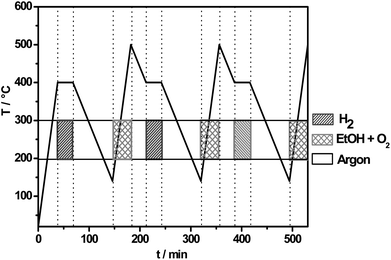 |
| | Fig. 16 Complete program of the temperature with the corresponding gas for the temperature programmed experiments. | |
Acknowledgements
Our work was supported through the Deutsche Forschungsgemeinschaft (DFG). TEM measurements at the Ernst-Ruska-Zentrum, Jülich, were made possible through the program ERC1-TUD. Dr L. Houben (Jülich) is acknowledged for technical assistance. Technical support of Dr K. Hofmann (XRD) and Dr A. Issanin (XPS) is acknowledged with gratitude.
References
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS.
-
C. Sanchez and P. Gomez-Romero, Functional Hybrid Materials, Wiley VCH, Weinheim, Germany, 2004 Search PubMed.
-
(a) L. Jiang and L. Gao, Carbon, 2003, 41, 2923–2929 CrossRef CAS;
(b) A. Fasi, I. Palinko, J. W. Seo, Z. Konya, K. Hernadi and I. Kiricsi, Chem. Phys. Lett., 2003, 372, 848–852 CrossRef CAS;
(c) B. Xue, P. Chen, Q. Hong, J. Lin and K. L. Tan, J. Mater. Chem., 2001, 11, 2378–2381 RSC;
(d) A. Chu, J. Cook, R. J. R. Heesom, J. L. Hutchison, M. L. H. Green and J. Sloan, Chem. Mater., 1996, 8, 2751–2754 CrossRef CAS.
-
(a) L. Cao, F. Scheiba, C. Roth, F. Schweiger, C. Cremers, U. Stimming, H. Fuess, L. Chen, W. Zhu and X. Qiu, Angew. Chem., Int. Ed., 2006, 45, 5315–5319 CrossRef CAS;
(b) S. Guo, S. Dong and E. Wang, J. Phys. Chem. C, 2008, 112, 2389–2393 CrossRef CAS.
-
(a) B. C. Satishkumar, A. Govindraj, J. Mofokeng, G. N. Subbanna and C. N. R. Rao, J. Phys. B: At., Mol. Opt. Phys., 1996, 29, 4925–4934 CrossRef CAS;
(b) L. Qu, L. Dai and E. Osawa, J. Am. Chem. Soc., 2006, 128, 5523–5532 CrossRef CAS.
-
(a) E. H. Espinosa, R. Ionescu, B. Chambon, G. Bedis, E. Sotter, C. Bittercourt, A. Felten, J. J. Pireaux, X. Correig and E. Llobet, Sens. Actuators, B, 2007, 127, 137–142 CrossRef;
(b) X. Wang, B. Xia, X. Zhu, J. Chen, S. Qiu and J. Li, J. Solid State Chem., 2008, 181, 822–827 CrossRef CAS;
(c) S. W. Lee and W. M. Sigmund, Chem. Commun., 2003, 780–781 RSC.
-
(a) J. Khanderi, R. C. Hoffmann, J. Engstler, G. Cherkashinin, J. Arras, J. J. Schneider and P. Claus, Chem.–Eur. J., 2010, 16, 2300–2308 CrossRef CAS;
(b) J. Khanderi, R. C. Hoffmann, A. Gurlo and J. J. Schneider, J. Mater. Chem., 2009, 19, 5039–5046 RSC.
-
(a) K. Akimoto, S. Ishizuka, M. Yanagita, Y. Nawa, G. K. Paul and T. Sakurai, Sol. Energy, 2006, 80, 715–722 CrossRef CAS;
(b) J. C. Park, J. Kim, H. Kwon and H. Song, Adv. Mater., 2009, 21, 803–807 CrossRef CAS;
(c) J. Zhang, J. Liu, Q. Peng, X. Wang and Y. Li, Chem. Mater., 2006, 18, 867–871 CrossRef CAS;
(d) Z. Yang, C.-K. Chiang and H.-T. Chang, Nanotechnology, 2008, 19, 025604 CrossRef;
(e) L. Huang, F. Peng, H. Yu and H. Wang, Solid State Sci., 2009, 11, 129–138 CrossRef CAS;
(f) L. Zhang, J. Li, Z. Chen, Y. Tang and Y. Yu, Appl. Catal., A, 2006, 299, 292 CrossRef CAS;
(g) T. Tsoncheva, R. Nickdov, S. Vankova and D. Mehandjiev, Can. J. Chem., 2003, 8, 1096 CrossRef;
(h) M. Scarselli, C. Sciletta, F. Tombolini, P. Castrucci, M. Diociaiuti, S. Casciardi, E. Gatto, M. Venanzi and M. De Crescenzi, J. Phys. Chem. C, 2009, 113, 5860–5864 CrossRef CAS.
-
(a) X. Zhang, G. Wang, W. Zhang, Y. Wei and B. Fang, Biosens. Bioelectron., 2009, 24, 3395–3398 CrossRef CAS;
(b) X. Wang, F. Zhang, B. Xia, X. Zhu, J. Chen, S. Qiu, P. Zhang and J. Li, Solid State Sci., 2009, 11, 655–659 CrossRef CAS;
(c) S. Dong, S. Zhang, L. Chi, P. He, Q. Wang and Y. Fang, Anal. Biochem., 2008, 381, 199–204 CrossRef CAS;
(d) A. Martinez-Ruiz and G. Alonso-Nunez, Mater. Res. Bull., 2008, 43, 1492–1496 CrossRef CAS;
(e) K. R. Reddy, B. C. Sin, C. H. Yoo, W. Park, K. S. Ryu, J. S. Lee, D. Sohn and Y. Lee, Scr. Mater., 2008, 58, 1010–1013 CrossRef CAS;
(f) Y. S. Luo, Q. F. Ren, J. L. Li, Z. J. Jia, Q. R. Dai, Y. Zhang and B. H. Yu, Nanotechnology, 2006, 17, 5836–5840 CrossRef;
(g) L. Zhang, J. Li, Z. Chen, Y. Tang and Y. Yu, Appl. Catal., A, 2006, 299, 292–297 CrossRef CAS;
(h) Y. Yu, L. L. Ma, W. Y. Huang, J. L. Li, P. K. Wong and J. C. Yu, J. Solid State Chem., 2005, 178, 1488–1494 CrossRef CAS;
(i) Y. Yu, L. L. Ma, W. Y. Huang, F. P. Du, J. C. Yu, J. G. Yu, J. B. Wang and P. K. Wong, Carbon, 2005, 43, 670–673 CrossRef CAS.
- T. S. Askgaard, J. K. Nørskov, C. V. Ovesen and P. Stoltze, J. Catal., 1995, 156, 229–242 CrossRef CAS.
- B. Lindstrom and L. J. Pettersson, Int. J. Hydrogen Energy, 2001, 26, 923–933 CrossRef CAS.
- A. Knop-Gericke, M. Hävecker, T. Schedel-Niedirg and R. Schlögl, Top. Catal., 2001, 15, 27–34 CrossRef CAS.
- E. Wachs and J. Madix, J. Catal., 1978, 53, 208 CrossRef CAS.
- E. Wachs and J. Madix, Appl. Surf. Sci., 1978, 1, 303 CrossRef CAS.
- I. Eswaramoorthi, V. Sundaramurthy and A. K. Dalai, Appl. Catal., A, 2006, 313, 22–34 CrossRef CAS.
-
(a) K. L. Hohn and Y.-C. Lin, ChemSusChem, 2009, 2, 927–940 CrossRef CAS;
(b) K. R. Reddy, B. C. Sin, C. H. Yoo, W. Park, K. S. Ryu., J. S. Lee, D. Sohn and Y. Lee, Scr. Mater., 2008, 58, 1010–1013 CrossRef CAS.
-
(a) N. Kanoun, M. P. Astier and G. M. Pajonk, Appl. Catal., 1991, 70, 225–236 Search PubMed;
(b) N. Kanoun, M. P. Astier and G. M. Pajonk, J. Mol. Catal., 1993, 79, 217–228 CrossRef CAS;
(c) Y.-J. Tu and Y.-W. Chen, Ind. Eng. Chem. Res., 1998, 37, 2618–2622 CrossRef CAS;
(d) Y.-J. Tu and Y.-W. Chen, Ind. Eng. Chem. Res., 2001, 40, 5889–5893 CrossRef CAS;
(e) F.-W. Chang, W.-Y. Kuo and K.-C. Lee, Appl. Catal., A, 2003, 246, 253–264 CrossRef CAS;
(f) F.-W. Chang, H.-C. Yang, L. S. Roselin, W.-Y. Kuo and K.-C. Lee, Appl. Catal., A, 2006, 304, 30–39 CrossRef CAS.
- Y.-J. Tu, C. Li and Y.-W. Chen, J. Chem. Technol. Biotechnol., 1994, 59, 141–147 CrossRef CAS.
- Y.-J. Tu, Y.-W. Chen and C. Li, J. Mol. Catal., 1994, 89, 179–190 CrossRef CAS.
- T. Y. Shin, S. H. Yoo and S. Park, Chem. Mater., 2008, 20, 5682–5686 CrossRef CAS.
- J. J. Schneider, R. C. Hoffmann, J. Engstler, O. Soffke, A. Issanin and A. Klyszcz, Adv. Mater., 2008, 20, 3383–3387 CrossRef CAS.
- A. Ahmad and I. D. Spencer, Can. J. Chem., 1961, 39, 1340–1359.
- Z. C. Orel, A. Anžlovar, G. Dražić and M. Žigon, Cryst. Growth Des., 2007, 7, 453–458 CrossRef.
- Z. Zhang, R. Liu, M. Zhao and Y. Qian, Mater. Chem. Phys., 2001, 71, 161–164 CrossRef CAS.
- P. E. Larson, J. Electron Spectrosc. Relat. Phenom., 1974, 4, 213 CrossRef CAS.
- Z. H. Gan, G. Q. Yu, B. K. Tay, C. M. Tan, Z. W. Zhao and Y. Q. Fu, J. Phys. D: Appl. Phys., 2004, 37, 81–85 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS.
- H. Werner, D. Herein, G. Schulz, U. Wild and R. Schlögl, Catal. Lett., 1997, 49, 109–119 CrossRef CAS.
-
(a) J. Ma, C. Park, N. M. Rodriguez and R. T. K. Baker, J. Phys. Chem. B, 2001, 105, 11994–12002 CrossRef CAS;
(b) R. T. K. Baker, J. Catal., 1982, 78, 473–476 CrossRef CAS.
- I. Böttger, T. Schedel-Niedirg, O. Timpe, R. Gottschall, M. Hävecker, T. Ressler and R. Schlögl, Chem.–Eur. J., 2000, 6, 1870–1876 CrossRef CAS.
- J. Agrell, M. Boutonnet and J. L. G. Fierro, Appl. Catal., A, 2003, 253, 213–223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: additional XRD and TG data. See DOI: 10.1039/c0nr00723d |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. ![X-Ray photoelectron spectrum of the O 1s region for [Cu2O/MWCNT] (solid line) and [Cu2O/ZnO/MWCNT] (dashed line) nanocomposites.](/image/article/2011/NR/c0nr00723d/c0nr00723d-f5.gif)