A facile synthetic route for the preparation of gold nanostars with magnetic cores and their reusable nanohybrid catalytic properties†
Xiumin
Miao
a,
Tingting
Wang
a,
Fang
Chai
ab,
Xiuli
Zhang
a,
Chungang
Wang
*a and
Wendong
Sun
*a
aCollege of Chemistry, Northeast Normal University, Changchun, 130024, P. R. China. E-mail: wangcg925@nenu.edu.cn
bCollege of Chemistry & Chemical Engineering, Harbin Normal University, Harbin, 150025, P. R. China
First published on 24th January 2011
Abstract
A facile synthetic route under mild conditions to the preparation of gold nanostars (GNSs) with Fe3O4 cores (Fe3O4@GNSs), possessing magnetization and tunable optical properties from the visible to near-infrared (NIR) region, was developed. Additionally, the resulting Fe3O4@GNSs described here show good catalytic activity for the reduction of potassium ferricyanide as a model reaction. Importantly, the catalyst, Fe3O4@GNSs, can be easily recycled with an external magnet and exhibits long-life, good reusability and stability. We also anticipated that Fe3O4@GNSs may provide a platform for broad potential diagnostic and therapeutic biomedical applications due to its magnetization and tunable optical properties from the visible to NIR region.
1. Introduction
Many attempts have been made to explore Fe3O4 nanoparticles (NPs) with superparamagnetism as the starting material to construct nanocrystals with a core-shell structure, due to their multiple potential applications in magnetic resonance imaging (MRI), magnetic recording devices, magnetically guided drug delivery, thermal therapy and magnetic separation.1–10 In particular, gold has been the preferred coating material due to its well-known catalytic activity, optical properties and chemical functionality.11–15 There have been several methods for the synthesis of Fe3O4/Au core-shell nanocrystals, including layer-by-layer electrostatic deposition,16 chemical reduction17,18 and reverse micelle method.19 However, there are few reports about gold nanostar (GNSs) coated Fe3O4 NPs, i.e. GNSs with Fe3O4 cores (designated as Fe3O4@GNSs). Very recently, Wei et al. reported a multistep synthesis of Fe3O4@GNSs via core-shell Fe3O4@Au as seeds that are prepared under solvothermal conditions using organic solvent (octyl ether) and under high temperature (190 °C).20 To overcome the above-mentioned disadvantages, we present, herein, a facile synthetic route to the preparation of Fe3O4@GNSs possessing magnetization and tunable optical properties from the visible to near-infrared (NIR) region at room temperature. Magnetically responsive Fe3O4@GNSs were prepared by a seeded growth method, starting from Au NP modified Fe3O4 NPs, through 11-mercaptoundecanoic acid (MUA) used as a linker, because the thiol groups of MUA promotes binding of small Au NPs, while the carboxylic acid group attaches to the Fe3O4 surface. Anisotropic growth was performed in micellar solutions of cetyltrimethylammonium bromide (CTAB) under mildly reducing conditions, resulting in the formation of Fe3O4@GNSs. Additionally, the resulting Fe3O4@GNSs described here exhibit good catalytic activity for the reduction of potassium ferricyanide. Importantly, the catalyst, Fe3O4@GNSs, can be easily recycled in the presence of an external magnet, and long-life and high reusability using an external magnetic field are also demonstrated. GNSs are multibranched NPs with sharp tips, which display interesting plasmonic properties and sharp tips of Fe3O4@GNSs could be utilized as magnetic surface-enhanced Raman scattering (SERS) tags for bioapplications. We also anticipated that Fe3O4@GNSs may provide a platform for the broad potential diagnostic and therapeutic biomedical applications due to its magnetization and tunable optical properties from the visible to NIR region.2. Experimental section
2.1 Materials
Fe3O4 NPs protected by oleylamine (OMA) and oleic acid (OA) were obtained as a gift from Ocean Nano Tech., tetramethylammonium hydroxide (10 wt % TMAOH), 11-mercaptoundecanoic acid (MUA, 99%), tetrakis (hydroxymethyl) phosphonium chloride (THPC), tetrachloroauric acid (HAuCl4·3H2O), AgNO3, L-ascorbic acid (AA), hexadecyltrimethylammonium bromide (CTAB) and sodium borohydride (NaBH4) were purchased from Sigma-Aldrich. Potassium ferricyanide (III) (K3Fe(CN)6) was supplied by Shanghai Chemical Corp. Deionized water was used for all experiments.2.2 Synthesis of MUA modified Fe3O4 NPs
Fe3O4 NPs protected by OMA/OA were dispersed in 5 mL of a 10 wt% TMAOH aqueous solution and sonicated for 10 min. Then the mixture was centrifuged at 8000 rpm for 10 min in order to remove excess surfactant (OMA/OA). The washing and sonicating processes were repeated 6 times to ensure as much surfactant is removed as possible. After removal of OMA/OA, the resulting Fe3O4 NPs were redispersed in 0.5 mL of deionized water. 100 μL of MUA was added into 0.5 mL of the above Fe3O4 NPs solution, which was then diluted with 2 mL of deionized water under mechanical stirring overnight. The resulting MUA modified Fe3O4 NPs were centrifuged at 8000 rpm for 10 min to further remove excess MUA. Then, the product was redispersed into deionized water and then collected with a magnet. This procedure was then repeated several times. Finally, the product was dispersed into 0.25 mL of deionized water for further use.2.3 Synthesis of Au NPs
Au NPs were synthesized by a previously reported method. Briefly, 0.1 mL of 1 M NaOH was added to 9 mL of deionized water, followed by the addition of 0.2 mL of THPC solution which was prepared by adding 12 μL of THPC (80%) to 1 mL of ultrapure water. After 5 min stirring, 0.3 mL of HAuCl4·3H2O (1 wt.%) was added rapidly. After further reaction for about 1 min with vigorously stirring, the color of the solution changed to dark brown. After further stirring for about 15 min, the resulting Au NPs with diameters of ca. 2 nm were stored at 4 °C for further use.2.4 Deposition of Au NPs onto the MUA modified Fe3O4 NPs (Fe3O4@Au NPs)
0.25 mL of MUA modified Fe3O4 NPs solution was added into 2 mL of Au NPs (THPC method) and 0.5 mL of ethanol which led to the formation of Fe3O4 NPs modified by the Au NPs. With overnight incubation under vortex mixing, high-density Au NPs could be attached on the surface of superparamagnetic Fe3O4 NPs using MUA as a linker. The resulting Fe3O4@AuNPs solution was centrifuged at 6000 rpm for 15 min. Then the product was redispersed into the water and separated by using the external magnetic field several times in order to remove the excess Au NPs which didn't attach to the surface of the MUA modified Fe3O4 NPs.2.5 Preparation of gold nanostars with Fe3O4 cores (Fe3O4@GNSs)
The mixture solution of 9.5 mL of 0.1 M CTAB, 0.4 mL of 10 mM HAuCl4·3H2O and 0.06 mL of 10 mM AgNO3 was prepared. Following the addition of 64 μL of AA with gentle shaking, the color of the solution changed from brown-yellow to colorless. Then 20 μL Fe3O4@AuNP seed solution was added. After the solution was mixed gently, it was kept at 27–30 °C without disturbance. The solution was faintly blue within 20 min, then the color changed from blue to dark blue gradually with the reaction time increasing up to 2 h. The resulting Fe3O4@GNSs product was separated and collected with a magnet, followed by washing with water at 8000 rpm three times within 10 min. Finally, the product was redispersed into water to form a homogeneous dispersion for further use.2.6 Catalytic reduction of K3Fe(CN)6
The reduction of K3Fe(CN)6 was carried out in a quartz cuvette and monitored using UV-vis spectroscopy at room temperature. In a typical reaction, 0.1 mL of 8 × 10−3 M K3Fe(CN)6 was added into 0.3 mL of Fe3O4@GNSs solution, followed by the rapid addition of 0.2 mL of 0.040 M ice-cold fresh NaBH4. The solution was then quickly subjected to UV-vis measurement. As the reaction proceeds, the color of the solution changed from yellow to colorless. To further investigate the reusability of the Fe3O4@GNSs as catalysts, the used Fe3O4@GNSs were separated from the solution with a magnet after the whole reduction process was complete. Similar to the above reduction process, the obtained Fe3O4@GNSs were redispersed in 0.3 mL of deionized water and mixed with 0.1 mL of 8 × 10−3 M K3Fe(CN)6 and 0.2 mL of 0.040 M ice-cold fresh NaBH4. The solution was measured using UV-vis spectroscopy quickly. The above-mentioned procedure was repeated 6 times.2.7 Characterization
UV-vis spectroscopy was performed with a U-3010 spectrophotometer (Hitachi, Japan). Transmission electron microscopy (TEM) was performed with a JEOL-100CX electron microscope. The magnetic measurement was carried out by using a superconducting quantum interference device magnetometer (SQUID MPMS XL-7) with fields up to 1.5 T.3. Results and discussion
Fig. 1A illustrates the facile strategy for the fabrication of Fe3O4@GNSs comprising a single, MUA-modified Fe3O4 NP decorated with GNSs. The strategy developed here for preparing bifunctional plasmonic magnetic Fe3O4@GNSs is to synthesize monodispersed Fe3O4 cores, attach discrete small Au NPs to the surface with bidentate ligands, MUA, which promotes binding of small Au NPs through the thiol group, while the carboxylic acid group attaches to the Fe3O4 surface.9 The resulting Fe3O4@AuNPs can be used as seeds for the formation of GNSs around Fe3O4 cores. Anisotropic growth of GNSs was carried out in the growth solution containing the mixture of CTAB, HAuCl4·3H2O, AgNO3 and L-ascorbic acid under mildly reducing conditions, leading to the formation of Fe3O4@GNSs, as shown in Fig. 1B. The Fe3O4 cores are no longer visible, because the GNSs shell was too thick to be electron transparent. | ||
| Fig. 1 (A) Schematic of synthetic procedure of gold nanostars with Fe3O4 cores (Fe3O4@GNSs) and (B) TEM image of Fe3O4@GNSs. MUA: 11-mercaptoundecanoic acid, CTAB: cetyltrimethylammonium bromide, AA: L-ascorbic acid. | ||
Monodispersed 25 nm Fe3O4 NPs stabilized with MUA by exchanging oleylamine (OMA) and oleic acid (OA) capping the surfaces of as-prepared Fe3O4 NPs were synthesized by a previously described procedure with slight modifications,21 as shown in Fig. 2A. About 2 nm gold seed clusters in water were prepared according to the approach previously reported by Duff et al. (Fig. 2B).22,23 The large difference in the radii of curvature of the Au NPs as seeds and the MUA modified Fe3O4 cores helps to achieve a high density of seed clusters on the surfaces of Fe3O4 NPs.21 The as-obtained Fe3O4 NPs were next transferred into water, achieved by removing excess OMA/OA surfactant with 10 wt% tetramethylammonium hydroxide (TMAOH). Then MUA was utilized to modify the above Fe3O4 NPs. The carboxylic acid groups of MUA can bind to the Fe3O4 NPs surfaces while the remaining thiol groups could be employed to attach small Au NPs through Au–S chemistry. Fig. 2C (a–c) show the UV-vis spectra of Fe3O4 NPs, Au NPs and Fe3O4@AuNPs, respectively. Fe3O4 and small Au NPs (ca. 2 nm) do not have any obvious absorbance peaks, as shown in Fig. 2C (a and b). However, the Fe3O4@AuNPs shows a clear surface plasmon resonance (SPR) band at 648 nm, indicating the formation of discrete Au clusters, and that the plasmonic response is attributed to interaction of Au clusters onto the Fe3O4 surface (Fig. 2C(c)), and not due to free Au NPs (Fig. 2C(b)). The photograph in Fig. 2D illustrates the effective magnetic separation of Fe3O4@AuNPs with an external magnet by removing unbound Au NPs in the solution. When a magnet was placed against the wall of a vial containing the suspension, the Fe3O4@GNSs were collected (Fig. 2D(b)). The remaining liquid exhibits a very slightly pink color, indicating that the bound together Au NPs and Fe3O4 NPs and unattached Au NPs have also been separated. The obtained Fe3O4@AuNPs was utilized as seeds for the seeded growth of GNSs in aqueous micellar solutions of cetyltrimethylammonium bromide (CTAB). GNSs growth onto the surface of Fe3O4 NPs as a function of time was first studied during an ongoing GNS synthesis by monitoring with UV-vis spectroscopy and, second, by imaging the morphological changes of GNS over time viaTEM. The growth of GNSs was monitored within 2 h by UV-vis spectroscopy, as shown in Fig. 3. The growth of consecutive GNSs is accompanied by a red shift and broadening of SPR bands from the visible to NIR region which is attributed to the formation of multibranch tips on the surface of Fe3O4 cores.
 | ||
| Fig. 2 TEM images of (A) MUA modified Fe3O4 NPs, (B) ca. 2 nm Au NPs, (C) UV-vis spectra of (a) Fe3O4 NPs, (b) Au NPs and (c) Fe3O4@AuNPs, respectively, (D) the photograph of Fe3O4@AuNPs as seeds for the fabrication of Fe3O4@GNSs in the (a) absence and (b) presence of an external magnet. | ||

To further elucidate the growth process of Fe3O4@GNSs, from the morphological comparison in the TEM images, as shown in Fig. 4A–E, the size of the Fe3O4@GNSs appears bigger and bigger and the morphology of the Fe3O4@GNSs becomes more and more “starlike” as the reaction time increases. The Fe3O4@GNSs became “starlike” obviously within about 1 h. The thickness of GNS shell gradually became much greater. Considering some factors, such as the size and the difficulty level of the magnetic separation, the Fe3O4@GNSs made within 1 h is much more suitable for applications because of not only the better morphology but also the relatively better magnetic property. In addition, it is well-known that the catalytic activity of a NP is strongly dependent on its size and shape, and consequently, the synthesis of colloidal NPs with well-controlled size and shape has become very important. Particularly, a smaller NP tends to show a higher catalytic activity due to its greater surface-to-volume ratio.24Fig. 4F shows that the Fe3O4@GNSs suspension is uniform and has a blue color before collection (Fig. 4F(a). Afterwards, it is clear, and Fe3O4@GNSs are clustered near the bottom edges of the cylindrical magnet, as shown in Fig. 4F(b). The magnetically responsive Fe3O4@GNSs were stable at room temperature, following the removal of excess CTAB.
 | ||
| Fig. 4 TEM images of monitoring the Fe3O4@GNS evolution over different times upon the addition of Fe3O4@AuNPs as seeds, (a) 20, (b) 30, (c) 40, (d) 65, (e) 105 min and (f) the photograph of Fe3O4@GNS suspension corresponding to sample (E) in the (a) absence and (b) presence of an external magnet. | ||
The Fe3O4@GNS solution’s color transitions as function of time, as depicted in Fig. 5 corresponding to the TEM images of A–E in Fig. 4. As the Fe3O4@AuNPs seeds grew and merged into a GNS, as shown in Fig. 4, the suspension became blue, and the SPR band red shifted to the NIR region (Fig. 3).
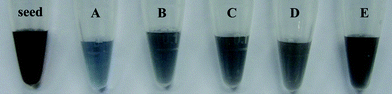 | ||
| Fig. 5 Photograph of Fe3O4@AuNP seed and as-synthesized Fe3O4@GNSs in water after different times, corresponding to the TEM images A–E in Fig. 4. | ||
Taken together, the UV-vis spectra and TEM images provide evidences with regard to the plausible growth mechanism of Fe3O4@GNSs. A high-resolution TEM (HRTEM) image of a well-formed Fe3O4@GNS is presented in Fig. S1.† The HRTEM image shows twin defects along the GNS tips which are similar to those found in gold nanorods (GNRs), where poor CTAB binding to twin defects leads to different degrees of exposure of certain facets for gold attachment and growth. The discrepancy of CTAB binding to different crystalline facets is thought to be a source of growth anisotropy due to GNS directed by CTAB surfactant.15,25 The formation of sharp tips of Fe3O4@GNSs is probably due to poor CTAB binding. The nucleation of growth anisotropy at multiple sites on the GNS could be due to a high defect density resulting in the growth of distinct branches with sharp tips by deposition of gold continuously. A more detailed growth mechanism is being studied through the investigation of the influence of the ratio between seeds and the growth solution, reaction temperature and so on.
Magnetic properties of Fe3O4 NPs and Fe3O4@GNSs are illustrated in Fig. 6. It can be seen from the hysteresis loops (Fig. 6a and b) that no remanence or coercivity was detected at room temperature, suggesting the superparamagnetic behavior of two samples due to their higher thermal fluctuation energy compared with their anisotropic energy.26 The saturation magnetization (SM) values of Fe3O4 NPs and Fe3O4@GNSs are 62.1 emu g−1 and 13.4 emu g−1, respectively, indicating that the significant decline of the SM value of Fe3O4@GNSs after decorating GNSs is due to the diamagnetic contribution of the GNSs surrounding the Fe3O4 cores.
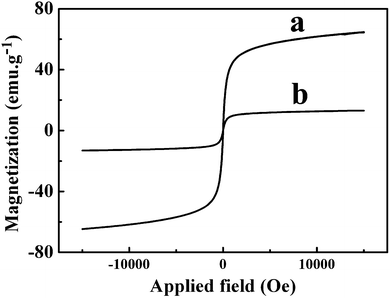 | ||
| Fig. 6 The magnetic hysteresis loops for (a) Fe3O4 NPs and (b) Fe3O4@GNSs recorded at 300 K from −15000 to +15000 Oe. | ||
It is noteworthy that recyclable, bifunctional magnetic NPs with high catalytic activity have received considerable interest because of their easy removal from mixtures.8–10,27,28 However, Fe3O4@GNSs are multibranched NPs with sharp tips prepared via a simple method which have not been studied as high-activity and recyclable catalyst. The present Fe3O4@GNSs have advantages such as a large catalytically active area, sharp Au tips on the surface and ease of recycling by magnetic separation. Herein, the reduction of K3[Fe(CN)6] with NaBH4 was selected as a model reaction for monitoring the catalytic activity and magnetic reusability of Fe3O4@GNSs. This reaction is chosen because it is a very “gentle” reaction that is catalyzed at room temperature. In addition, the whole reaction process is rapid, and can be monitored spectroscopically.29,30 The reduction of K3[Fe(CN)6] by NaBH4 in aqueous solution can be written as:31,32
| BH4− + 8Fe(CN)63− + 3H2O → H2BO3− + 8Fe(CN)64− + 8H+ |
Fig. 7A and B show the UV-vis absorption of the reduction reaction of K3[Fe(CN)6] in the absence and presence of catalyst, Fe3O4@GNSs. The light yellow aqueous K3[Fe(CN)6] solution shows absorption at 420 nm. After the addition of NaBH4, the intensity of absorption gradually decreased due to the formation of K4[Fe(CN)6]. The whole reduction process took about 12 h (Fig. 7A). However, after the addition of a small amount of Fe3O4@GNSs, the absorption peak at 420 nm significant decreased and the reaction process totally completed within 1 min in the first run, indicating the high catalytic activity of Fe3O4@GNSs, as shown in Fig. 7B.
![(A) UV-vis spectra of the reduction of K3[Fe(CN)6] solution upon the addition of NaBH4 recorded at different times from 1 min to 12 h at 25 °C, [[Fe(CN)6]3−] = 8 × 10−3 M, [BH4−] = 0.04 M in the absence of catalyst, Fe3O4@GNSs, (B) reaction in the presence of Fe3O4@GNSs within 1 min: (a) only 8 × 10−3 M K3[Fe(CN)6], (b) 8 × 10−3 M K3[Fe(CN)6] solution upon the addition of 0.04 M NaBH4 with Fe3O4@GNSs and (c) only Fe3O4@GNSs, (C) and (D) show the performance of the catalyst recycled twice, (E) reduction of K3[Fe(CN)6] in 6 successive cycles and magnetic separation, (F) the photograph of the solution of K3[Fe(CN)6] and NaBH4 (a) before and (b) after the addition of Fe3O4@GNSs and (c) magnetic separation of Fe3O4@GNSs in the presence of an external magnet.](/image/article/2011/NR/c0nr00704h/c0nr00704h-f7.gif) | ||
| Fig. 7 (A) UV-vis spectra of the reduction of K3[Fe(CN)6] solution upon the addition of NaBH4 recorded at different times from 1 min to 12 h at 25 °C, [[Fe(CN)6]3−] = 8 × 10−3 M, [BH4−] = 0.04 M in the absence of catalyst, Fe3O4@GNSs, (B) reaction in the presence of Fe3O4@GNSs within 1 min: (a) only 8 × 10−3 M K3[Fe(CN)6], (b) 8 × 10−3 M K3[Fe(CN)6] solution upon the addition of 0.04 M NaBH4 with Fe3O4@GNSs and (c) only Fe3O4@GNSs, (C) and (D) show the performance of the catalyst recycled twice, (E) reduction of K3[Fe(CN)6] in 6 successive cycles and magnetic separation, (F) the photograph of the solution of K3[Fe(CN)6] and NaBH4 (a) before and (b) after the addition of Fe3O4@GNSs and (c) magnetic separation of Fe3O4@GNSs in the presence of an external magnet. | ||
To further investigate the reusability, the Fe3O4@GNSs were separated using a magnet from the catalytic reaction solution. The catalyst exhibits similar catalytic performance in the second and third runs, as examples, respectively, (Fig. 7C and D). As shown in Fig. 7E, the catalyst, Fe3O4@GNSs, can be successfully recycled and still keep their high activity after reusing in 6 successive reactions. From the 4th to the 6th run, conversions of about 100% was achieved within 5 min, indicating a slight increase of reaction time. We found that the Fe3O4@GNSs remain colloidally stable, and no aggregation takes place after being magnetically separated 6 times, as indicated by TEM image (Fig. S2†), further suggesting their excellent stability and long life. The photograph in Fig. 7F shows that the solution is yellow before catalytic reaction and becomes transparent after catalytic reaction (Fig. 7F a and b). When the reaction was complete, the catalyst were quickly separated from the solution using a magnet (Fig. 7c) and transferred to another new reaction system for the next cycle of catalysis. Importantly, the synthesis of Fe3O4@GNSs can be extended to other Pt and Pd-based GNNs to obtain recoverable high activity catalysts for different catalytic reactions.
4. Conclusion
In summary, we have demonstrated a facile synthetic route to the fabrication of gold nanostars with Fe3O4 cores, Fe3O4@GNSs, under mild reaction conditions. In addition, the use of Fe3O4@GNSs as a novel catalyst for the reduction of potassium ferricyanide in the presence of NaBH4 was also investigated. The results indicated that as-prepared Fe3O4@GNSs nanohybrids exhibit good performance as a magnetically recyclable catalyst with no significant loss of catalytic activity after reusing in 6 successive reactions. Additionally, GNSs are multibranched NPs with sharp tips, which display interesting plasmonic properties and the sharp tips of Fe3O4@GNSs could be utilized as magnetic surface-enhanced Raman scattering (SERS) tags for bioapplications. We also anticipated that Fe3O4@GNSs may provide a platform for broad potential in diagnostic and therapeutic biomedical applications due to its magnetization and tunable optical properties from the visible to NIR region.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Project No. 20703009, 21037031), Fundamental Research Funds for the Central Universities, the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20070200021) and the Department of Science and Technology of Jilin Province (20082103).References
- L. Y. Wang, H. Y. Park, S. I. Lim, M. J. Schadt, D. Mott, J. Luo, X. Wang and C. J. Zhong, J. Mater. Chem., 2008, 18, 2629 RSC.
- Y. W. Jun, J. H. Lee and J. W. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122 CrossRef CAS.
- M. K. Yu, Y. Y. Jeong, J. Park, S. Park, J. W. Kim, J. J. Min, K. Kim and S. Y. Jon, Angew. Chem., Int. Ed., 2008, 47, 5362 CrossRef CAS.
- F. M. Kievit, O. Veiseh, C. Fang, N. Bhattarai, D. Lee, R. G. Ellenbogen and M. Q. Zhang, ACS Nano, 2010, 4, 4587 CrossRef CAS.
- P. Howes, M. Green, A. Bowers, D. Parker, G. Varma, M. Kallumadil, M. Hughes, Warley, A. Brain and R. Botnar, J. Am. Chem. Soc., 2010, 132, 9833 CrossRef CAS.
- Z. C. Xu, Y. L. Hou and S. H. Sun, J. Am. Chem. Soc., 2007, 129, 8698 CrossRef CAS.
- Y. H. Wei, R. Klajn, A. O. Pinchu and B. A. Grzybowski, Small, 2008, 4, 1635 CrossRef CAS.
- S. J. Guo, S. J. Dong and E. K. Wang, Chem.–Eur. J., 2009, 15, 2416 CrossRef CAS.
- Y. P. Wu, T. Zhang, Z. H. Zheng, X. B. Ding and Y. X. Peng, Mater. Res. Bull., 2010, 45, 513 CrossRef CAS.
- Y. H. Deng, Y. Cai, Z. K. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Y. Zhao, J. Am. Chem. Soc., 2010, 132, 8466 CrossRef CAS.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721 CrossRef CAS.
- C. J. Xu, J. Xie, D. Ho, C. Wang, N. Kohler, E. G. Walsh, J. R. Morgan, Y. E. Chin and S. H. Sun, Angew. Chem., Int. Ed., 2008, 47, 173 CrossRef CAS.
- C. Kim, E. C. Cho, J. Y. Chen, K. H. Song, L. Au, C. Favazza, Q. Zhang, C. M. Cobley, F. Gao, Y. N. Xia and L. V. Wang, ACS Nano, 2010, 4, 4559 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957 CrossRef CAS.
- C. L. Nehl, H. W. Liao and J. H. Hafner, Nano Lett., 2006, 6, 683 CrossRef CAS.
- M. Spasova, V. Salgueirino-Maceira, A. Schlachter, M. Hilgendorff, M. Giersig, L. M. Liz-Marzan and M. Farle, J. Mater. Chem., 2005, 15, 2095 RSC.
- L. Y. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. H. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593 CrossRef CAS.
- S. F. Chin, K. S. Iyer and C. L. Raston, Cryst. Growth Des., 2009, 9, 2685 CrossRef CAS.
- S. J. Cho, A. M. Shahin, G. J. Long, J. E. Davies, K. Liu, F. Grandjean and S. M. Kauzlarich, Chem. Mater., 2006, 18, 960 CrossRef CAS.
- Q. S. Wei, H. M. Song, A. P. Leonov, J. A. Hale, D. Oh, Q. K. Ong, K. Ritchie and A. Wei, J. Am. Chem. Soc., 2009, 131, 9728 CrossRef CAS.
- J. Lim, A. Eggeman, F. Lanni, R. D. Tilton and S. A. Majetich, Adv. Mater., 2008, 20, 1721 CrossRef CAS.
- D. G. Duff and A. Baiker, Langmuir, 1993, 9, 2301 CrossRef CAS.
- C. C. Huang, Z. S. Yang, K. H. Lee and H. T. Chang, Angew. Chem., Int. Ed., 2007, 46, 1 CrossRef.
- J. Zeng, Q. Zhang, J. Y. Chen and Y. N. Xia, Nano Lett., 2010, 10, 30 CrossRef CAS.
- C. G. Khoury and T. V. Dinh, J. Phys. Chem. C, 2008, 112, 18849 CAS.
- D. Chen, J. J. Li, C. S. Shi, X. W. Du, N. Q. Zhao, J. Sheng and S. Liu, Chem. Mater., 2007, 19, 3399 CrossRef CAS.
- D. K. Yi, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 2459 CrossRef CAS.
- S. J. Guo, L. Wang, S. J. Dong and E. K. Wang, Chem.–Eur. J., 2009, 15, 2416 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2004, 126, 7194 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343 CrossRef CAS.
- S. Carregal-Romero, J. Perez-Juste, P. Herves, L. M. Liz-Marzan and P. Mulvaney, Langmuir, 2010, 26, 1271 CrossRef CAS.
- T. Freund, J. Inorg. Nucl. Chem., 1959, 9, 246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM image of Fe3O4@GNSs after running for 6 times by magnetic separation. See DOI: 10.1039/c0nr00704h |
| This journal is © The Royal Society of Chemistry 2011 |