Biomineral nanoparticles are space-filling†
Li
Yang
a,
Christopher E.
Killian
bc,
Martin
Kunz
d,
Nobumichi
Tamura
d and
P. U. P. A.
Gilbert
*b
aEarth Science Division, Lawrence Berkeley National Lab, Berkeley, CA 94720, USA
bDepartment of Physics, University of Wisconsin, Madison, WI 53706, USA. E-mail: pupa@physics.wisc.edu
cDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720, USA
dAdvanced Light Source, Lawrence Berkeley National Lab, Berkeley, CA 94720, USA
First published on 17th November 2010
Abstract
Sea urchin biominerals have been shown to form from aggregating nanoparticles of amorphous calcium carbonate (ACC), which then crystallize into macroscopic single crystals of calcite. Here we measure the surface areas of these biominerals and find them to be comparable to those of space-filling macroscopic geologic calcite crystals. These biominerals differ from synthetic mesocrystals, which are invariably porous. We propose that space-filling ACC is the structural precursor for echinoderm biominerals.
Introduction
In this study we address the question: are echinoderm biominerals mesocrystals? The answer appears to be no, based on previous evidence of amorphous precursors and the data presented here on the space-filling of biominerals. But let us first define the terms biominerals, mesocrystals, porosity, and space filling.Marine biominerals are most frequently composites of organic molecules and calcite or aragonite (CaCO3). The fascinating morphology of mollusk shells, corals, and echinoderm biominerals along with the remarkable mechanical performances of these materials have motivated extensive studies of the mechanisms leading to their formation.1,2 Several biominerals have been shown to form from nanoparticulate carbonates, yet the mature biominerals are macroscopic single crystals of calcite.3,4 As a consequence, a concept has emerged that such biominerals are best described as so-called mesocrystals, formed by the assembly of pre-formed units.5,6 There is evidence from studies of natural and synthetic systems that nanoparticles can be oriented and epitaxially joined during aggregation or self-assembly, potentially providing a mechanism for the formation of the complex-morphology single-crystalline biominerals observed. However, imaging studies of marine biominerals reveal these materials to be virtually non-porous, that is, completely space-filling. No self-assembly method in the laboratory has been shown to achieve this. Here, we directly analyze several echinoderm biominerals to determine if there are indeed mesocrystals present, and then discuss the implications of porosity and space-filling in biomineralization.
A mesocrystal is a three-dimensional arrangement of perfectly co-oriented nanocrystals. Mesocrystals were first discovered and then comprehensively reviewed by Cölfen.7 They are usually produced synthetically, by several mechanisms reviewed in detail by Meldrum and Cölfen.6 In most synthetic mesocrystals, nanoparticles adhere to one another by a mechanism of oriented attachment. This mechanism was first discovered by the Banfield group in a synthetic system,8 and subsequently in a bacterially-induced biomineral.9 For synthetic mesocrystals to form, the nanoparticles must be crystalline first, and subsequently specific crystalline facets “dock”, that is, come in contact with one another and minimize energy by fusing together.9,10 In other synthetic mesocrystals, corners instead of facets of crystalline nanoparticles attach to one another.11 In our view the three key aspects of all synthetic mesocrystals are: (I) the nanoparticles are crystalline before aggregating, (II) the resulting assembly of nanoparticles is highly co-oriented, and (III) invariably highly porous.12,13
Sea urchin (echinoderm) biominerals are model systems to explore the fundamental mechanisms of biomineral crystal formation. These biominerals include embryonic larval spicules, as well as adult spines, tests, and teeth. Sea urchin spicules, spines, and teeth have been shown to form via amorphous precursor phases (ACC),4,14–17 while spicules and the polycrystalline matrix of the sea urchin teeth have also been shown to form via aggregation of amorphous nanoparticles.4,16 Sea urchin spines have been confirmed to behave as single crystals of calcite, even with high-resolution backscattered electron diffraction,18 despite the fact that they formed from an amorphous precursor phase.15 Whether the ACC initially deposited is subdivided into nanoparticles has not been established for spines. Forming sea urchin spicules4 and teeth19 have been reported to be composed of calcite nanoparticles, with sizes on the order of 50 nm,4,15,17 yet in visible light microscopy with crossed-polarizers, X-ray diffraction, and spectromicroscopy, they behave as macroscopic single4,15 or double crystals,16,20 respectively. This means that the nanoparticles in sea urchin spicules and teeth are highly co-oriented. This unusual property of biominerals, being subdivided into nanoparticles yet having long-range co-orientation, raises several interesting questions, which have not yet been addressed. Do the nanoparticles retain their separation in space once the biomineral is fully formed, and mature? Do they join one another through mineral bridges, as synthetic mesocrystals often do7? Or, do they fuse into a space-filling continuum and no longer have the large surface areas characteristic of nanoparticles?
In this work we address these questions with a technique that is well-established in the nanoparticle community, but which, surprisingly, has not been widely used to analyze natural biominerals: surface area measurements using the Brunauer-Emmett-Teller (BET) method.21,22 Briefly, during BET analysis, the mineral or biomineral sample is first ground finely into a powder, sieved, and then outgassed extensively to remove moisture or gasses adsorbed onto the particle surfaces. Subsequently a noble gas is adsorbed onto the particle surfaces, by physisorption, not chemisorption. The amount of gas adsorbed is determined quantitatively and accurately. These measurements yield the precise surface area of the powder particles or nanoparticles in units of m2/g. If the material measured is a porous aggregate of nanoparticles, or a solid space-filling single crystal, the measured surface area is large or small, respectively. To our knowledge, the only previous BET study of biominerals examined porosity artificially induced in silica diatoms shells.23 BET, however, has been used extensively to reveal that synthetically produced mesocrystals, containing organic polymers and calcium carbonate, have a large surface area, in the range of 260 m2/g, and are therefore highly porous.24,25
Spheres with identical radii of any size can only fill 74.05% of the space in the hexagonal close-packing (HCP) configuration, that is, spheres packed as cannon balls in a stack, while using multiple radii space-filling greater than 75% can be achieved.26,27Do nanoparticles in sea urchin biominerals match, or even exceed the HCP space-filling capability, as they assemble their nanoparticles? In synthetic nanoparticle systems, complete space-filling is difficult to achieve, in fact, to our knowledge, only the Murray group is capable of densely packing nanoparticles, using various binary nanoparticle systems of metals (Au, Pd, or Ag) and oxides, or sulfides, or selenides.27,28 The surface areas have not been measured by BET in these particular systems (personal communication by C. B. Murray), however most nanoparticle systems of comparable sizes have been observed to have surface areas on the order of 250–320 m2/g.29–31
BET analysis provides the opportunity to examine natural biominerals and investigate if they are or are not mesocrystals. Here we use BET to analyze sea urchin adult spines and teeth, as well as embryonic spicules, and compare the results with a geologic calcite crystal, which fills 100% of the space and is therefore expected to have the smallest surface area. Particular emphasis is placed on sea urchin spines, which are a macroscopically porous material, comprised of the sponge-like calcite structure known as “the stereom”.32 We also compared these data with those from the sea urchin tooth, which is comprised of large calcite crystals 1–10 μm-thick, ∼1000 μm-long plates and fibers32 cemented together by nanoparticles of Mg-rich calcite termed “the polycrystalline matrix”.
Results
In Fig. 1A, we show an SEM image of a fractured sea urchin spine, which shows the spine's well-known sponge-like stereom internal morphology. Despite this non-faceted, elaborate morphology, the entire spine diffracts as a single crystal of calcite and is often cited as an example of a co-oriented natural “mesocrystal”.7,33Fig. 1A also shows that the fracture figure of a spine is not one of the usual cleavage planes in geologic calcite, rather it is a conchoidal surface, which broke like an amorphous glass (see arrow in the figure for example). In Fig. 1B and C we show SEM images of a sea urchin spicule and a tooth also from S. purpuratus. The calcite mineral of these structures is known to be highly co-oriented and to fracture conchoidally. The presence of intra-crystalline proteins causes this conchoidal fracturing, as first demonstrated by the Addadi-Weiner group,34 and subsequently by others.![Biominerals from the sea urchin Strongylocentrotus purpuratus. [A] Scanning electron micrograph of a fractured sea urchin spine. Notice the conchoidal fracture figure of the central stereom and the external sectors. The arrow indicates the glass-like conchoidal fracture of a sector. [B] SEM micrograph of a tri-radiate spicule extracted from 36 h-old larvae of the sea urchin, and bleached. Despite its morphology and the rounded surface, the spicule diffracts as a single crystal of calcite. [C] Scanning electron micrograph of a bleached sea urchin tooth at its grinding tip. This is an oblique view. The tooth is comprised of plates and fibers, which diffract as single crystals of calcite, and a polycrystalline matrix that fills the space between the plates and fibers as the tooth matures. This polycrystalline matrix is comprised of Mg-rich calcite nanoparticles.](/image/article/2011/NR/c0nr00697a/c0nr00697a-f1.gif) | ||
| Fig. 1 Biominerals from the sea urchin Strongylocentrotus purpuratus. [A] Scanning electron micrograph of a fractured sea urchin spine. Notice the conchoidal fracture figure of the central stereom and the external sectors. The arrow indicates the glass-like conchoidal fracture of a sector. [B] SEM micrograph of a tri-radiate spicule extracted from 36 h-old larvae of the sea urchin, and bleached. Despite its morphology and the rounded surface, the spicule diffracts as a single crystal of calcite. [C] Scanning electron micrograph of a bleached sea urchin tooth at its grinding tip. This is an oblique view. The tooth is comprised of plates and fibers, which diffract as single crystals of calcite, and a polycrystalline matrix that fills the space between the plates and fibers as the tooth matures. This polycrystalline matrix is comprised of Mg-rich calcite nanoparticles. | ||
We used micro-X-ray diffraction to examine the nanoparticulate nature of the sea urchin tooth polycrystalline matrix. Fig. 2 shows one of the X-ray diffraction patterns obtained from the polycrystalline matrix in the tooth of S. purpuratus. Diffraction from a single crystal of calcite generates a distinct pattern of isolated single spots or reflections. The pattern in Fig. 2, instead, shows many clusters of multiple reflections, indicating that the crystals in the polycrystalline matrix are small, and closely but not perfectly co-oriented. Each cluster of reflections reveals that the nanoparticles are ∼10 nm in size, and their orientation spread is on the order of 1 degree.16 This is, to our knowledge, the only biomineral system in which the nanoparticles are and remain distinct from one another in the mature biomineral, and they are also the smallest of any biomineral structure thus far analyzed, although it is not yet known what keeps the nanoparticles separate in the polycrystalline matrix.
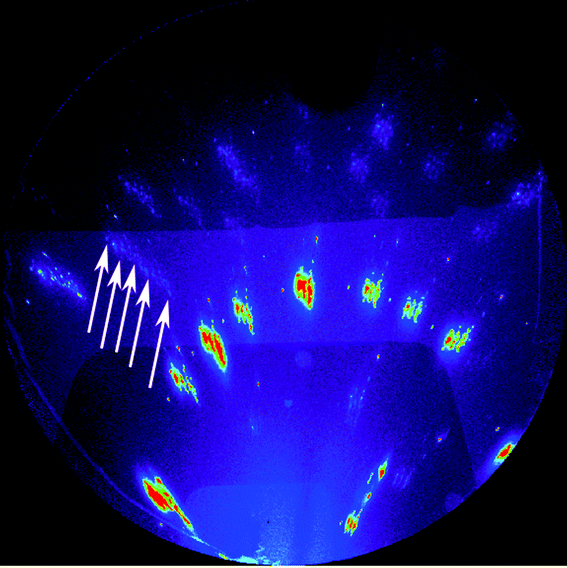 | ||
| Fig. 2 Diffraction pattern of the polycrystalline matrix nanoparticles at the tip of the sea urchin tooth. Instead of single-spot reflections we observe clusters of faint spots. The arrows indicate a few of these spots in one cluster. The subdivision of each calcite reflection into weak small spots indicates that individual diffracting grains are smaller than the illuminating X-ray beam spot size. Based on the size of the X-ray beam (1 μm2), we estimate that the grain size is on the order of 10 nm. The close proximity of the spots in each cluster shows that these nanoparticles are highly co-oriented. The width in 2θ of the spread gives the orientation spread of the nanoparticles, which is 1° in full width at half maximum, and 4° in maximum spread. | ||
In Table 1 we show the results of BET analysis from samples similar to those in Fig. 1, in addition to spines from four other sea urchin species, and calcite. After the initial analysis of these biominerals, we noticed the similarity of the results among the spicules, spines and teeth. The surface areas of all these echinoderm biominerals are not significantly different from that of calcite, indicating that they fill space as much as a single crystal of geologic calcite. This surprising result deserved further investigation.
| Sample description | Mass analyzed | Surface area measured by BET ± 5% error |
|---|---|---|
| Geologic calcite | 0.0798 g | 1.23 ± 0.06 m2/g |
| Sp larval spicules (72 h) | 0.0082 g | 1.26 ± 0.06 m2/g |
| Sp tooth (with nanoparticles) | 0.0158 g | 2.39 ± 0.12 m2/g |
| Sp adult spines | 0.0497 g | 1.24 ± 0.06 m2/g |
| Pl adult spines | 0.1967 g | 2.28 ± 0.11 m2/g |
| Et adult spines | 0.1511 g | 1.82 ± 0.09 m2/g |
| Ap adult spines | 0.1105 g | 0.84 ± 0.04 m2/g |
| Lv adult spines | 0.1020 g | 1.08 ± 0.05 m2/g |
The only differences observed in Table 1 are across the spines from different species, which may reflect small differences in their porosity. However, they are significantly less porous than separate nanoparticles, or synthetic mesocrystals, which exhibit surface areas consistently in the range 200–300 m2/g. Given these surprising results, we decided to extensively bleach the sea urchin tooth sample, because, as learned from diffraction (Fig. 2), the sea urchin teeth are indeed subdivided into nanoparticles, which are interspersed with the large single crystals of plates and fibers.16 A small measured surface area (2.39 ± 0.12 m2/g) implies that either the nanoparticles are fused together, albeit mis-oriented, or that they are connected by organic molecules, which themselves fill the inter-nanoparticle space. Many proteins have been identified in the organic matrix of the sea urchin tooth35,36 and may play a role in maintaining the small nanoparticle size in the polycrystalline matrix of the tooth. Bleach oxidizes and dissolves proteins, which are then removed by washing, and is therefore expected to significantly increase the surface area measured.
In Table 2 we present the BET results before and after extensive bleaching of the sea urchin tooth powder. After extensive bleaching, the surface area of the sea urchin tooth more than doubled, strongly suggesting that organics, indeed, were interspersed with the nanoparticles. Dissolving the organics away with the bleach revealed more of the nanoparticles' surface area. The increase in surface area is significant, but not as large as one would have expected from a sample made up of pure 10 nm particles (∼100 m2/g). However, the tooth samples contained a significant proportion of large (micron-sized) plates and fiber structures that diffract as single crystals.
| Sample description | Mass analyzed | Surface area measured by BET ±5% error |
|---|---|---|
| Sp teeth (with nanoparticles) | 0.0158 g | 2.39 ± 0.12 m2/g |
| Sp teeth (with nanoparticles) extensively bleached | 0.0992 g | 6.76 ± 0.34 m2/g |
| Sp adult spines | 0.0497 g | 1.24 ± 0.06 m2/g |
| Sp adult spines, extensively bleached | 0.2792 g | 1.72 ± 0.09 m2/g |
Interestingly, the extensive bleach treatment did not significantly affect the surface area measured in S. purpuratus spines, as also shown in Table 2. This resistance to bleach treatment indicates that sea urchin spines are single crystals, not subdivided into nanoparticles held together by proteins.
Discussion
The coherence length of synthetic calcite is 500 nm,37 while the coherence lengths of sea urchin single-crystal biominerals are significantly shorter. Berman et al. showed that for the sea urchin tooth fibers the coherence length is 200 nm, for adult spines it is 150 nm, and for larval spicules it is 100 nm.37 The co-orientation in these biominerals is within 0.1°.37 It is difficult to correlate these diffraction data with the surface areas measured here, because what separates nano-scale units, or domains with a certain coherence length in a single crystal is unknown. Nevertheless, the comparison of BET results from geologic calcite and biominerals enables some definite conclusions. The BET results presented here show that all the echinoderm biominerals behave as macroscopic single crystals of calcite rather than a porous aggregate of nanoparticles separated in space.Porosity in the sponge-like stereom of the sea urchin spines is at the macroscopic level—on the 10-μm scale (Fig. 1A). This stereom porosity is at a much larger scale than the nanoscopic (<2 nm) or mesoscopic (2 nm–100 nm) scale. Whether or not the sea urchin spine is initially formed by nanoparticles of ACC, is not yet known. However, the fully formed mature spine does not show evidence of subdivision into nanoparticles. Even after extensive bleaching we did not observe the subdivision of spines into calcite nano-bricks suggested by Oaki and Imai.38 These observations may indicate that organic molecules do not surround the nano-brick structures observed by Oaki and Imai. Our results are consistent across five different sea urchin species (Table 1), so we believe they are reliable and confirmed.
In contrast to the sea urchin spine, the sea urchin tooth after extensive bleaching increases in surface area. It is known that nanoparticles exist in the sea urchin tooth3 and they behave as separate entities in diffraction (Fig. 2). Dispersed, 10-nm nanoparticles would be expected to give a surface area greater than 100 m2/g. In the sea urchin tooth, however, the polycrystalline matrix nanoparticles are not dispersed but tightly aggregated. The observation that after extensive bleaching the surface area only increased by a factor of ∼2, is probably due to the co-existence of the large-single-crystal components—the plates and the fibers—which are not affected by bleaching. The fact that the surface area increased, however, provides evidence that organic molecules are in part responsible for the tight aggregation of the nanoparticles in the polycrystalline matrix. Fusion of crystals, minimizing surface energy by forming mineral-to-mineral interfaces must have occurred, even though the nanoparticles exhibit an angular spread on the order of 1°.
We conclude from the BET data that biominerals are space-filling. They fill space more efficiently than current synthetic nanoparticle systems, and as efficiently as macroscopic single crystals of geologic calcite. This observation was unexpected, and warrants a direct critical analysis of natural biominerals vis-à-vis synthetic mesocrystals.
As mentioned above, synthetic mesocrystals7 form by the mechanism of oriented attachment8 of crystalline nanoparticles. Many biominerals, perhaps most of them, form following a different mechanism. Amorphous precursor phases have been identified in forming biominerals from different phyla: echinoderms,4,14–16 mollusks,39,40 crustaceans,41–46 annelids47–49 and chordates.50,51Crystallizationvia amorphous precursor phases, therefore, may be a general strategy in biomineralization.16 These amorphous nanoparticles aggregate, fill space, and subsequently crystallize by a mechanism of secondary nucleation as described by Killian et al.16 This mechanism has been first observed in sea urchin spicules,4 and then also in the polycrystalline matrix of the sea urchin tooth,16 in which the pattern of crystallinity propagation through the amorphous phase could be directly imaged.
The different formation mechanisms between natural biominerals and synthetic mesocrystals, however, may not be received as significant, if one adopts a very broad definition of mesocrystallinity, as proposed by Cölfen.7 Can a biomineral that forms from ACC be considered a mesocrystal? According to the definition of mesocrystals given above (I), the answer is no, because mesocrystallinity requires that the nanoparticles be crystalline, not amorphous, before aggregation. Should one adopt a different definition the answer may of course differ. However one answers this question, the crux of the matter is: which measurable characteristics do synthetic mesocrystals and natural biominerals have in common?
To address this question one may examine: (i) the perfect co-orientation in mesocrystals6 which is not always observed in biominerals, when analyzed with high-resolution methods;16,37,52–54 (ii) the much higher concentration of organics in carbonate mesocrystals7 than in biominerals;55,56 (iii) the presence of facets in mesocrystals,6 which is conspicuously absent in biominerals; (iv) finally, the space-filling reported here, which is far greater for biominerals than for mesocrystals. The surface areas are in fact in the 1–2 m2/g range for the biominerals examined here, and >250 m2/g for mesocrystals.24,25,31,57
There may be a structural advantage to forming biominerals through amorphous precursors. The amorphous minerals can be morphed into the intricate shapes of the final biomineral, which exhibit the curved surfaces of spicules, spines, and teeth, which are under the direct control of the organism and are presumably optimized for their support, sheltering, grinding and deterring functions. We propose that there is another structural advantage of the ACC precursor phase: space-filling. It is obvious that space can be better filled by amorphous nanoparticles, which have no facets, are initially hydrated4 and thus able to flow and fill space. Filling space with amorphous mineral precursors is also fast and efficient.5 A non-porous, space-filling solid is structurally and mechanically stronger than a porous one. In this scenario, space-filling provides greater robustness and resistance to fracture to the biomineral and consequently affords a competitive advantage to the host organism.
Materials and methods
Calcite
Part of a 10 × 10 × 10 cm3 rhombohedron of crystalline geologic calcite from Beetown, WI (Bernie's Rock Shop, Madison, WI) was crushed in an agate mortar and pestle, sieved through a 75-μm copper grid to eliminate the larger powder grains, and analyzed with BET.Sea urchin spines
Entire sea urchins from 5 species (Strongylocentrotus purpuratus, Paracentrotus lividus, Arbacia punctulata, Eucidaris tribuloides, Lytechinus lividus) were bleach-treated and their spines collected. The smallest spines were collected for scanning electron microscopy analysis, while the largest spines were collected for BET analysis. The dry spines were crushed in an agate mortar and pestle, then sieved through a 75-μm copper grid. For the S. purpuratus spines only, after crushing into a powder, part of the spines were bleached while vortexing for 10 min, and centrifuged at 20,000 g for 5 min. Bleaching and centrifuging was repeated twice, to remove as much of the intra-crystalline or inter-nanoparticle organics as possible, while retaining the mineral nanoparticles or nano-bricks, in case they existed in the original spine. The extensively bleached powder was then rinsed twice in ethanol, centrifuged, and dried.Sea urchin teeth
The jaw apparatuses (Aristotle's Lanterns) of adult sea urchins from one species, Strongylocentrotus purpuratus, were removed and placed in 5.25% NaClO for approximately 20 min, to remove the organic components. After removing the lanterns from the bleach, individual teeth were dissected from the pyramids of the lanterns and placed in 2 ml microcentrifuge tubes. The teeth were then washed twice with water and twice with 100% ethanol. The teeth were then allowed to air dry for 15 min. For SEM analysis the teeth were analyzed directly, and without coating. For BET analysis S. purpuratus teeth were further dissected with a scalpel under a stereomicroscope. The tip ends of 50 teeth (≤5 mm from the tip) were separated from the rest of the teeth, crushed all together in a mortar and pestle, and sieved through a 75 μm copper grid to eliminate the larger powder grains. At this point part of the powder was analyzed with BET, and the rest was bleached and centrifuged twice to further remove intra-crystalline or inter-nanoparticle organics, while retaining the polycrystalline matrix nanoparticles, presumably dispersed into the bleach solution. Again, bleaching was done for 10 min while vortexing, and centrifuging at 20,000 g for 5 min. The extensively bleached powder was then rinsed twice in ethanol, centrifuged, and dried.Sea urchin spicules
S. purpuratus embryos were grown in filtered natural sea water containing gentamycin (20 mg L−1) at 15 °C, following established methods.58 Seventy-two hours after fertilization, at the pluteus stage, embryos were disrupted using a Polytron homogenizer, the spicules were collected by centrifugation, extracted with SDS, and bleached with 3.5% NaOCl. The spicules were washed with CaCO3 saturated DD-H2O, rinsed with ethanol and acetone, and kept at −80 °C for several months, then at room temperature for one week until the BET measurement.Scanning electron microscopy
SEM analysis was done at the Electron Microscopy Lab, University of California at Berkeley. The environmental SEM used for imaging the sea urchin spines and teeth is a Hitachi TM1000 SEM, in the back-scattered electron (BSE) imaging mode. This environmental SEM does not require sample coating. All samples were simply mounted on carbon tape and imaged at a fixed voltage of 15 kV. The sea urchin spicules were imaged using a Hitachi S-5000 Scanning electron microscope. The spicules were mounted on carbon tape and sputter coated with 1 nm Au/Pd using a Tousimis Sputter Coater. The spicules were imaged using a secondary electron detector at a fixed voltage of 20 kV.Micro-X-ray diffraction
S. purpuratus spines and teeth were analyzed either in reflection or transmission mode, using 1 μm × 1 μm pink beam of photon energy 5–22 keV, from a super-bend source at the Advanced Light Source, beamline 12.3.2. The horizontal μ-beam illuminated the sample, mounted at 45°, and the X-Ray CCD detector (MAR133 from MAR, now Rayonix) was vertical and on top, with the active area at 89 mm distance from the sample. The sample was mounted on and x-y-z scanning stage, so either maps or single (Laue) XRD patterns could be acquired in the reflection mode, with an exposure time of 1 s. Fig. 2 was taken in the transmission mode, with the CCD detector at 70° from the downstream direction of the beam. Only the tooth tip, was positioned in the beam path. This is the thinnest, most distal portion of the tooth tip, which contains only polycrystalline matrix nanoparticles protruding from the last of the tooth plates.BET analysis
Brunauer-Emmett-Teller surface area analysis was done using an Autosorb®−1 instrument (Quantachrome Instruments, Florida) at the Lawrence Berkeley National Laboratory. All biominerals samples were treated identically. They were crushed in a mortar and pestle, and sieved through a 75 μm copper mesh to eliminate the larger powder grains. The powder collected thereafter was placed into a 6 mm glass cell, then outgassed in vacuum for at least 18 h at 50 °C, to remove any moisture or gasses adsorbed onto the powder particle surfaces. The sample cells were then stabilized at liquid nitrogen temperature (77.350 K) and measured for BET surface area with krypton gas. Multiple-point BET surface area measurements (corresponding to P/Po ratios of 0.050, 0.075, 0.100, 0.150, and 0.200) were performed for each sample to determine its specific surface area. The measurement and corresponding specific surface area calculation were done using the instrument software Quantachrome AS1Win™(version 2.01).Acknowledgements
We thank Benjamin Gilbert, Christopher B. Murray, and Fred H. Wilt for useful discussions. The diffraction data were acquired at the LBNL-ALS, supported by DOE under contract DE-AC02-05CH11231. This work was supported by DOE award DE-FG02-07ER15899, NSF award CHE/DMR-0613972, and UW-Hamel Award to PUPAG.References
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, Oxford, 1989 Search PubMed.
- K. Mann, I. Weiss, S. André, H. J. Gabius and M. Fritz, Eur. J. Biochem., 2001, 267, 5257–5264 CrossRef.
- Y. R. Ma, S. R. Cohen, L. Addadi and S. Weiner, Adv. Mater., 2008, 20, 1555–1559 CrossRef CAS.
- Y. Politi, R. A. Metzler, M. Abrecht, B. Gilbert, F. H. Wilt, I. Sagi, L. Addadi, S. Weiner and P. U. P. A. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17362–17366 CrossRef CAS.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS.
- H. Cölfen and M. Antonietti, Mesocrystals and nonclassical crystallization, John Wiley & Sons, Chichester, UK, 2008 Search PubMed.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549–1557 CrossRef CAS.
- J. F. Banfield, S. A. Welch, H. Zhang, T. T. Ebert and R. L. Penn, Science, 2000, 289, 751–754 CrossRef CAS.
- A. P. Alivisatos, Science, 2000, 289, 736–737 CrossRef CAS.
- A. N. Kulak, P. Iddon, Y. T. Li, S. P. Armes, H. Colfen, O. Paris, R. M. Wilson and F. C. Meldrum, J. Am. Chem. Soc., 2007, 129, 3729–3736 CrossRef CAS.
- R. Q. Song, H. Colfen, A. W. Xu, J. Hartmann and M. Antonietti, ACS Nano, 2009, 3, 1966–1978 CrossRef CAS.
- R. Q. Song and H. Colfen, Adv. Mater., 2010, 22, 1301–1330 CrossRef CAS.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461–465 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS.
- C. E. Killian, R. A. Metzler, Y. T. Gong, I. C. Olson, J. Aizenberg, Y. Politi, L. Addadi, S. Weiner, F. H. Wilt, A. Scholl, A. Young, S. N. Coppersmith and P. U. P. A. Gilbert, J. Am. Chem. Soc., 2009, 131, 18404–18409 CrossRef CAS.
- A. V. Radha, T. Z. Forbes, C. E. Killian, P. U. P. A. Gilbert and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16438–16443 CrossRef CAS.
- C. Moureaux, A. Perez-Huerta, P. Compere, W. Zhu, T. Leloup, M. Cusack and P. Dubois, J. Struct. Biol., 2010, 170, 41–49 CrossRef CAS.
- Y. R. Ma, S. R. Cohen, L. Addadi and S. Weiner, Adv. Mater., 2008, 20, 1555–1559 CrossRef CAS.
- Y. R. Ma, B. Aichmayer, O. Paris, P. Fratzl, A. Meibom, R. A. Metzler, Y. Politi, L. Addadi, P. U. P. A. Gilbert and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6048–6053 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of porous solids and powders: surface area, pore size and density, Springer, Dordrecht, 2006 Search PubMed.
- C. E. Fowler, Y. Hoog, L. Vidal and B. Lebeau, Chem. Phys. Lett., 2004, 398, 414–417 CrossRef CAS.
- T. X. Wang, H. Colfen and M. Antonietti, J. Am. Chem. Soc., 2005, 127, 3246–3247 CrossRef.
- T. P. Wang, M. Antonietti and H. Colfen, Chem.–Eur. J., 2006, 12, 5722–5730 CrossRef CAS.
- Z. Y. Chen, J. Moore, G. Radtke, H. Sirringhaus and S. O'Brien, J. Am. Chem. Soc., 2007, 129, 15702–15709 CrossRef CAS.
- J. Chen, X. C. Ye and C. B. Murray, ACS Nano, 2010, 4, 2374–2381 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef.
- Z. L. Wang, S. A. Harfenist, I. Vezmar, R. L. Whetten, J. Bentley, N. D. Evans and K. B. Alexander, Adv. Mater., 1998, 10, 808–812 CrossRef CAS.
- J. W. Zhu, W. G. Zhang, H. Z. Wang, X. J. Yang, L. D. Lu and X. Wang, Chinese Journal of Inorganic Chemistry, 2004, 20, 863–867 Search PubMed.
- D. S. Wang, T. Xie, Q. Peng and Y. D. Li, J. Am. Chem. Soc., 2008, 130, 4016–4022 CrossRef CAS.
- P. U. P. A. Gilbert and F. H. Wilt, Molecular aspects of biomineralization of the echinoderm endoskeleton, in Molecular Biomineralization: Aquatic Organisms Forming Extraordinary Materials, ed. W.E.G. Mueller, 2011, Elsevier, in press. Search PubMed.
- N. A. J. M. Sommerdijk and H. Cölfen, Materials Research Society Bullettin, 2010, 35, 116–119 Search PubMed.
- A. Berman, L. Addadi and S. Weiner, Nature, 1988, 331, 546–548 CrossRef CAS.
- K. Mann, A. J. Poustka and M. Mann, Proteome Sci., 2008, 6, 33 CrossRef.
- K. Mann, A. J. Poustka and M. Mann, Proteome Sci., 2010, 8, 6 CrossRef.
- A. Berman, J. Hanson, L. Leiserowitz, T. F. Koetzle, S. Weiner and L. Addadi, Science, 1993, 259, 776–779.
- Y. Oaki and H. Imai, Small, 2006, 2, 66–70 CrossRef CAS.
- I. M. Weiss, N. Tuross, L. Addadi and S. Weiner, J. Exp. Zool., 2002, 293, 478–491 CrossRef CAS.
- N. Nassif, N. Pinna, N. Gehrke, M. Antonietti, C. Jager and H. Cölfen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12653–12655 CrossRef CAS.
- S. Raz, O. Testeniere, A. Hecker, S. Weiner and G. Luquet, Biol. Bull., 2002, 203, 269–274 CrossRef CAS.
- F. Neues, A. Ziegler and M. Epple, CrystEngComm, 2007, 9, 1245–1251 RSC.
- K. E. Chave, J. Geol., 1954, 62, 266–283 CrossRef CAS.
- A. P. Vinogradov, The Elementary Chemical Composition of Marine Organisms. Sears Foundation for Marine Research II, Yale University Press, New Haven, 1953 Search PubMed.
- Y. Levi-Kalisman, S. Raz and S.e. a. Weiner, J. Chem. Soc., Dalton Trans., 2000, 3977–3982 RSC.
- R. Dillaman, S. Hequembourg and M. Gay, J. Morphol., 2005, 263, 356–374 CrossRef.
- C. Darwin, The Formation of Vegetable Mould, Through the Action of Worms, with Observations on Their Habits, John Murray, London, 1881 Search PubMed.
- M. R. Lee, M. E. Hodson and G. N. Langworthy, Mineral. Mag., 2008, 71, 257–261 CrossRef.
- I. Gago-Duport, M. J. I. Briones and J. B. Rodriguez, et. al, J. Struct. Biol., 2008, 162, 422–435 CrossRef.
- J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748–12753 CrossRef CAS.
- E. Beniash, R. A. Metzler, R. S. K. Lam and P. U. P. A. Gilbert, J. Struct. Biol., 2009, 166, 133–143 CrossRef CAS.
- C. Hedegaard and H.-R. Wenk, J. Molluscan Stud., 1998, 64, 133–136 CrossRef.
- D. Chateigner, C. Hedegaard and H.-R. Wenk, J. Struct. Geol., 2000, 22, 1723–1735 CrossRef.
- P. U. P. A. Gilbert, R. A. Metzler, D. Zhou, A. Scholl, A. Doran, A. Young, M. Kunz, N. Tamura and S. N. Coppersmith, J. Am. Chem. Soc., 2008, 130, 17519–17527 CrossRef CAS.
- P. E. Hare and P. H. Abelson, Carnegie Institution of Washington Yearbook, 1965, 64, 223–234 Search PubMed.
- F. Wilt, J. Struct. Biol., 1999, 126, 216–226 CrossRef CAS.
- R. Z. Wang, J. Am. Ceram. Soc., 1998, 81, 1037–1040 CAS.
- K. R. Foltz, N. L. Adams and L. L. Runft, Meth Cell Biol, 2004, 74, 40–71 Search PubMed.
Footnote |
| † This article was submitted as part of a Themed Issue on Crystallization and Formation Mechanisms of Nanostructures. Other papers on this topic can be found in issue 11 of vol. 2 (2010). This issue can be found from the Nanoscale homepage [http://www.rsc.org/nanoscale] |
| This journal is © The Royal Society of Chemistry 2011 |