Facile synthesis of an up-conversion luminescent and mesoporous Gd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :Er3+@nSiO2@mSiO2 nanocomposite as a drug carrier
:Er3+@nSiO2@mSiO2 nanocomposite as a drug carrier
Zhenhe
Xu
ab,
Chunxia
Li
a,
Ping'an
Ma
*a,
Zhiyao
Hou
a,
Dongmei
Yang
ab,
Xiaojiao
Kang
ab and
Jun
Lin
*a
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022
bGraduate University of the Chinese Academy of Sciences, Beijing, 100049, P. R. China. E-mail: mapa675@ciac.jl.cn; jlin@ciac.jl.cn
First published on 22nd November 2010
Abstract
In this paper, we report the facile synthesis of a bifunctional inorganic nanocomposite which is composed of core-shell structured mesoporous silica coated with up-conversion Gd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Er3+ particles. X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), N2adsorption/desorption, and photoluminescence (PL) spectra were used to characterize the samples. The results indicate that the nanocomposite with general 50 nm shell thickness and 300 nm core size shows typical ordered mesoporous characteristics (2.3 nm) and has spherical morphology with smooth surface and narrow size distribution. The bifunctional system shows unique green up-conversion emission under 980 nm NIR laser excitation even after loading with drug molecules. In addition, biocompatibility tests on L929 fibroblast cells using an MTT assay reveals low cytotoxicity of the system. Drug release tests suggest that the nanocomposite has a controlled drug release property with ibuprofen (IBU) as the model drug. Interestingly, the up-conversion emission intensity of the bifunctional carrier increases with the released amount of model drug, thus allowing the release process to be monitored and tracked by the change of up-conversion luminescence intensity. This composite can potentially act as a functional drug carrier system.
:Er3+ particles. X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), N2adsorption/desorption, and photoluminescence (PL) spectra were used to characterize the samples. The results indicate that the nanocomposite with general 50 nm shell thickness and 300 nm core size shows typical ordered mesoporous characteristics (2.3 nm) and has spherical morphology with smooth surface and narrow size distribution. The bifunctional system shows unique green up-conversion emission under 980 nm NIR laser excitation even after loading with drug molecules. In addition, biocompatibility tests on L929 fibroblast cells using an MTT assay reveals low cytotoxicity of the system. Drug release tests suggest that the nanocomposite has a controlled drug release property with ibuprofen (IBU) as the model drug. Interestingly, the up-conversion emission intensity of the bifunctional carrier increases with the released amount of model drug, thus allowing the release process to be monitored and tracked by the change of up-conversion luminescence intensity. This composite can potentially act as a functional drug carrier system.
1. Introduction
The combination of nanotechnology and molecular biology has developed into an emerging area: nanobiotechnology.1 So the design and synthesis of multifunctional nanomedical platforms that integrate suitable multiple nanomaterials with different properties into a single nanosystem provides an unparalleled opportunity for the simultaneous diagnostics and therapy of diseases.2 In particular, lanthanide-doped up-conversion nanoparticles (UCNPs) that convert a near-infrared excitation into visible emissions have been developed as a new class of luminescent optical labels in biological assays and medical imaging.3,4 Compared with down-conversion materials, the up-conversion materials can emit higher-energy visible photons after being excited by lower-energy near-infrared (NIR) photons. Meanwhile, excitation with the NIR only results in a very weak autofluorescence background because the UV-excitable biomolecules (biological tissues and fluorescent drug molecules) that interfered with normal phosphor luminescence can no longer be excited by NIR radiation.5,6In recent years, much attention has been paid to developing new drug storage/release systems with many advantages compared with the conventional forms of dosage, such as enhanced bioavailability, greater efficiency, lower toxicity, controlled release, and so on.7,8 In general, an efficient drug delivery system can not only deliver the therapeutic drugs to the targeted cells or tissues but also maintain the optimum concentration and rational toxicity of drugs in precise sites of the organs, which can improve therapeutic efficiency and reduce toxicity.9 Up to now, a large variety of systems have been successfully employed as means of sustained/controlled drug delivery for their improved drug loading efficiency and degradable property, such as biodegradable polymers,7a hydroxyapatite (HAp),10,11calcium phosphate cement (CFC),12,13 xerogels,14 hydrogels,15,16 and so on. Among different storage/release systems, ordered mesoporous silica materials have gained enhanced interest with particular attention as drug storage and release hosts due to their unique properties including stable mesoporous, tunable pore size, high specific surface area, and well modifiable surface.17 Furthermore, there has also been considerable attention in modifying the host matrices to either increase the magnetism or introduce new functional groups.18 The combination of mesoporous silica with luminescent functional groups to form core–shell structured composite is undoubtedly of special interest in diagnostic analysis,19enzyme immobilization,20 bioseparation,21 and controlled drug release22 based on their unique visible luminescence, low cytotoxicity, good biocompatibility, and mesoporous properties. Thus, the design of core-shell-structured nanocomposites with these special features should be highly promising in biomedical fields.
In this article, we present a facile procedure for synthesizing a bifunctional nanocomposite composed of ordered mesoporous silica encapsulating Gd2O3:Er3+ up-conversion luminescence nanoparticles with core-shell structures. The drug storage/release properties were also investigated on this system based on their mesoporous and luminescent properties using ibuprofen (IBU) as a model drug.
2. Experimental section
2.1. Synthesis of monodisperse Gd0.99Er0.01(OH)CO3 colloid spheres
All the chemical agents used in this experiment were of analytical grade and used directly without further purification. The monodisperse colloid spheres of Gd0.99Er0.01(OH)CO3 were prepared via a urea-based homogeneous precipitation process.23 A total of 0.75 mL of Gd0.99Er0.01(NO3)3 (1 M) and 1.5 g of urea [CO(NH2)2] were dissolved in deionized water. The total volume of the solution was about 50 mL. The above solution was first homogenized under magnetic stirring at room temperature for 2 h. The resultant solution was then reacted at 90 °C for 2 h in the oil bath. The obtained suspension was separated by centrifugation and collected after washing with deionized water several times.2.2. Synthesis of Gd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Er3+@nSiO2@mSiO2 nanocomposite
:Er3+@nSiO2@mSiO2 nanocomposite
In a typical procedure, as-prepared Gd0.99Er0.01(OH)CO3 (0.10 g) nanoparticles were treated with ethanol by ultrasonication for 30 min. Subsequently, the treated nanoparticles were separated by centrifugation, and then well dispersed in a mixture of ethanol (40 mL), deionized water (10 mL), and concentrated ammonia aqueous solution (28 wt %, 0.5 mL). Tetraethoxysilane (TEOS, 0.015 g) was then added dropwise to the solution. After stirring for 6 h, the products were separated by centrifugation and washed with ethanol and water, and then redispersed in a mixed solution containing cetyltrimethylammonium bromide (CTAB) (0.15 g), deionized water (40 mL), concentrated ammonia aqueous solution (28 wt %, 0.6 mL), and ethanol (30 mL). The resulting solution was stirred for 30 min. TEOS (0.2 g) was then added dropwise to the solution with stirring. After stirring for a further 6 h, the products were collected and separated by centrifugation, washed with ethanol and water several times, and dried in air at 80 °C for 24 h and calcined at 550 °C for 6 h. Finally, the CTAB-removed product was denoted as Gd2O3:Er3+@nSiO2@mSiO2.
2.3. The biocompatibility of the core-shell structured Gd2O3:Er3+@nSiO2@ mSiO2 nanocomposite
To evaluate the biocompatibility of the Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite, an MTT cell assay was used on the Vero cell line. MTT is a standard test for screening the toxicity of biomaterials and is carried out in accordance with ASTM standards. This method is based on the formation of dark red formazan by the metabolically active cells after their exposure to MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). Briefly, plate 5000–6000 L929 fibroblast cells in 200 μL media per well in a 96 well plate, leave 8 wells empty for blank controls. Incubate (37 °C, 5% CO2) overnight to allow the cells to attach to the wells. The Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite was sterilized by ultraviolet irradiation for 2 h, and then serial dilutions of the nanocomposite with concentrations of 0.5, 1.25, 2.5, 5, 10, and 20 mg mL−1 were added to the culture wells to replace the original culture medium and incubated for another 24 h in 5% CO2 at 37 °C. 5 mg ml−1 stock solution of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was prepared in PBS and this stock solution (20 μL) was added to each well containing a different amount of the Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite. The plate was covered with aluminium foil for protection from light and incubated at 37 °C for 4 h. During this period viable cells reduce MTT to formazan pigment, which can be dissolved in dimethyl sulfoxide (DMSO)/isopropanol. After incubation, 100 μL of acidified isopropanol was added to each well, and place on a shaking table, 150 rpm for 5 min, to thoroughly mix the formazan into the solvent. The absorbance of the suspension was recorded under a microplate reader at 570 nm.
2.4. Preparation of drug storage/delivery system
The drug storage/release system using the core-shell structured Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite as a carrier was prepared according to the previous reports.24Ibuprofen (IBU) was selected as the model drug. Typically, 0.2 g of the core-shell structured Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite sample was added into 30 mL hexane solution with an IBU concentration of 60 mg mL−1 at room temperature, and soaked for 24 h with stirring in a vial that was sealed to prevent the evaporation of hexane. The IBU-loaded sample was separated by centrifugation, and then dried under vacuum at 60 °C for 24 h, and denoted as IBU–Gd2O3:Er3+@nSiO2@mSiO2.
The in vitro delivery of IBU was performed by immersing 0.2 g of the sample in the release media of simulated body fluid (SBF) with slow stirring under the immersing temperature of 37 °C. The ionic composition of the as-prepared SBF solution was similar to that of human body plasma with a molar composition of 142.0/5.0/2.5/1.5/147.8/4.2/1.0/0.5 for Na+/K+/Ca2+/Mg2+/Cl−/HCO3−/HPO42−/SO42− (pH = 7.4). The ratio of SBF to adsorbed IBU was kept at 1 mL mg−1. At selected time intervals, a sample (0.5 mL) was removed and immediately replaced with an equal volume of fresh SBF. The solution removed was properly diluted and the amount of ibuprofen present was monitored at 222 nm using a UV-vis spectrophotometer.
2.6. Characterization
The X-ray diffraction (XRD) patterns of the samples were carried out on a D8 Focus diffractometer (Bruker) with use of Cu-Kα radiation (λ = 0.15405 nm). The morphology and composition of the samples were inspected using a scanning electron microscope (FE-SEM; XL 30, Philips). Transmission electron microscopy (TEM) micrographs were obtained from an FEI Tecnai G2 S-twin transmission electron microscope with a field emission gun operating at 200 kV. Nitrogen adsorption/desorption analysis was measured using Micromeritics ASAP 2020 M apparatus. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method using the data between 0.05 and 0.35. The UV–vis adsorption spectral values were measured on a TU-1901 spectrophotometer. The UC emission spectra of were obtained using a 980 nm laser from an OPO (optical parametric oscillator, Continuum Sunlite, USA) as the excitation source and detected by a R955 (HAMAMATSU) from 400 to 700 nm. All the measurements were performed at room temperature.3. Results and discussion
The procedure for the fabrication of Gd2O3:Er3+@nSiO2@mSiO2 bifunctional inorganic nanocomposite is illustrated in Scheme 1. First, uniform Gd0.99Er0.01(OH)CO3 particles were synthesized by a general urea-based homogeneous precipitation method. Scanning electron microscopy (SEM) images for the pure Gd0.99Er0.01(OH)CO3 confirm that the as-prepared sample consists of monodisperse spheres with a mean particle size of 330 nm and smooth surface (Fig. 1A, B). These particles are non-aggregated with narrow size distribution. Fig. 1C and D show typical transmission electron microscopy (TEM) images of monodisperse Gd0.99Er0.01(OH)CO3 spheres, which have a relatively smooth surface and an average diameter of about 330 nm. Second, a mesoporous silica shell offers many advantages as the framework for the multifunctional nanoparticles. The mesoporous silica shell (50 nm) was synthesized around the Gd0.99Er0.01(OH)CO3 spheres by a modification of the procedures described by Zhao et al.25 In this procedure, Gd0.99Er0.01(OH)CO3 particles were first modified with nonporous SiO2 layer through a modified Stöber procedure,26 resulting in the formation of the silica-Gd0.99Er0.01(OH)CO3 composite with a non-porous silica layer of 5 nm in thickness (denoted as Gd0.99Er0.01(OH)CO3@nSiO2) (Fig. 2). Subsequently, cetyltrimethylammonium bromide (CTAB) was selected as the organic template for the formation of the outer mesoporous silica layer on Gd:Er(OH)CO3@nSiO2. After heat treatment at 550 °C for 6 h, the CTAB was removed, accompanied by the decomposition of Gd0.99Er0.01(OH)CO3 and formation of Gd2O3:Er3+, and the final sample was designated as Gd2O3:Er3+@nSiO2@mSiO2. The FESEM images of samples with different magnifications show that the spheres are very uniform in both size and shape (Fig. 3A, B). The morphological and structural features of the Gd2O3:Er3+@nSiO2@mSiO2 spheres were further examined by TEM. From Fig. 3C, D, the core-shell structure can be clearly distinguished due to the different electron penetrability between the cores and shells. The Gd2O3:Er3+ cores are black spheres with an average size of about 300 nm, and the silica shell shows gray color with an average thickness of 50 nm. Notably, quasi hexagonal mesopore channels are clearly found to be perpendicular to the spheres' surface (Fig. 3E).
 | ||
| Scheme 1 The formation process of multifunctional Gd2O3:Er3+@nSiO2@mSiO2 nanocomposites. | ||



The wide-angle XRD patterns of the Gd0.99Er0.01(OH)CO3, Gd2O3:Er3+, and Gd2O3:Er3+@nSiO2@mSiO2 are displayed in Fig. 4. The XRD pattern of the precursor shows two broad bands at 30° and 45° (Fig. 4A), which indicates that the sample is amorphous. After thermal treatment at 550 °C for 6 h, the cubic phase Gd2O3 [space group: Ia ![[3 with combining macron]](https://www.rsc.org/images/entities/i_char_0033_0304.gif) (206)] with lattice constant a = 1.0790 nm is obtained, suggesting that Gd2O3 particles were well retained in the silica matrix (Fig. 4C). Fig. 5 shows the low-angle XRD pattern of Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite. A strong (100) peak in the low-angle XRD pattern reveals an ordered 2D mesopore symmetry, which suggests the short range ordering character of the sample. The N2 adsorption/desorption isotherm of Gd2O3:Er3+@nSiO2@mSiO2 is shown in Fig. 6. As shown, the sample exhibits typical VI-type isotherms with H1-hysteresis loops, which are usually related with hexagonal cylindrical channels. The Brunauer–Emmett–Teller (BET) surface area of the sample and pore volume are measured to be 365 m2g−1 and 0.25 cm3g−1, respectively. Moreover, the pore-size distribution shows a narrow apex centered at 2.3 nm (inset in Fig. 6). This result further indicates that the as-prepared Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite has mesopore channels.
(206)] with lattice constant a = 1.0790 nm is obtained, suggesting that Gd2O3 particles were well retained in the silica matrix (Fig. 4C). Fig. 5 shows the low-angle XRD pattern of Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite. A strong (100) peak in the low-angle XRD pattern reveals an ordered 2D mesopore symmetry, which suggests the short range ordering character of the sample. The N2 adsorption/desorption isotherm of Gd2O3:Er3+@nSiO2@mSiO2 is shown in Fig. 6. As shown, the sample exhibits typical VI-type isotherms with H1-hysteresis loops, which are usually related with hexagonal cylindrical channels. The Brunauer–Emmett–Teller (BET) surface area of the sample and pore volume are measured to be 365 m2g−1 and 0.25 cm3g−1, respectively. Moreover, the pore-size distribution shows a narrow apex centered at 2.3 nm (inset in Fig. 6). This result further indicates that the as-prepared Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite has mesopore channels.
 | ||
| Fig. 4 The wide-angle XRD patterns of (A) the Gd0.99Er0.01(OH)CO3 particles, (B) Gd2O3:Er3+ sample, and (C) Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite. | ||
 | ||
| Fig. 5 The low-angle XRD pattern of the Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite. | ||
 | ||
| Fig. 6 N2 adsorption–desorption isotherms and mesopore size distribution (the inset) of the synthesized Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite. | ||
The up-conversion (UC) luminescence spectra of Gd2O3:Er3+@nSiO2@mSiO2 and IBU–Gd2O3:Er3+@nSiO2@mSiO2 samples under 980 nm NIR laser excitation are shown in Fig. 7. The Gd2O3:Er3+@nSiO2@mSiO2 sample with bright green emission under the excitation of 980 nm NIR laser (inset in Fig. 7). Two bands in the green emission region maximized at 539 and 562 nm are assigned to the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions of Er3+, respectively, and a weak band that appears at about 661 nm is attributed to the 4F9/2 → 4I15/2 transition of Er3+ (Fig. 7, red line).27 The UC mechanism of the up-converted green emission of Er3+ is shown in Fig. 8. The excitation wavelength of 980 nm matches the absorption transition between the ground state, 4I15/2, and the excited level 4I11/2 (GSA) of Er3+. After first-level excitation, the same wavelength laser pumps the excited atom from the 4I11/2 to the 4F7/2 level (ESA). Subsequent nonradiative relaxation populates the 2H11/2, 4S3/2, and 4F9/2 levels. Finally, radiant transitions from these levels yield the emissions at 539 and 562 nm (2H11/2, 4S3/2 → 4I15/2) (most strong) and at 661 nm (4F9/2 → 4I15/2), respectively. Particularly, the characteristic emission peaks are still obvious except for the decrease of intensity for IBU–Gd2O3:Er3+@nSiO2@mSiO2 (Fig. 7, black line).
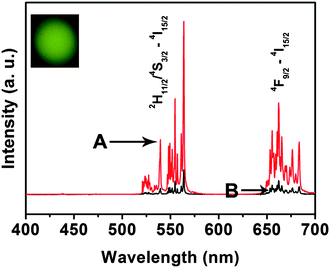 | ||
| Fig. 7 Up-conversion emission spectra of (A) Gd2O3:Er3+@nSiO2@mSiO2, and (B) IBU–Gd2O3:Er3+@nSiO2@mSiO2. | ||
 | ||
| Fig. 8 Energy level diagram of Er3+ and the proposed UC emission mechanism. | ||
In order to evaluate the biocompatibility of the storage and release properties, an MTT assay was performed on the nanocomposite. This method is based on the formation of dark red formazan by the metabolically active cells after their exposure to MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). Cell viability is directly proportional to the amount of formazan produced, monitored by the absorbance at 570 nm. In Fig. 9A we can see that more than 90% L929 fibroblast cells viability was observed under the high concentration range (0.5–20 mg mL−1), thus indicative of the good biocompatibility of the nanocomposite in all dosages. Fig. 9B and C show the inverted microscopy images of the cells grown in the presence of the nanocomposite, which reveal all cells got spread and began to proliferate, and their density increased gradually. The above experimental results indicate that this nanocomposite is non-toxic and promising for biomedical applications.
 | ||
| Fig. 9 The biocompatibility of (A) the Gd2O3:Er3+@nSiO2@mSiO2 nanocomposite was analyzed using MTT assay. L929 fibroblast cells were incubated with the nanocomposite for 24 h. Microscopic images of Vero cells. Incubation time: (B) 0 h; (C) 24 h. | ||
The ordered mesoporous materials (MSMs) can be used as a drug delivery host because MSMs are non–cytotoxic, and are able to carry a high payload of guest molecules within the nanopores.28 Here we test for the drug storage and release properties of this system as a candidate of drug carriers, and ibuprofen was selected as a model drug, which has been extensively investigated for sustained and controlled drug delivery due to its short biological half-life (2 h), good pharmacological activity and the suitable molecule size (1.0 × 0.6 nm). During the loading and release process, the IBU molecules can be adsorbed onto the surface of mesoporous materials in the impregnation process and liberated by a diffusion-controlled mechanism. The OH groups on the surface should be the reactive sites that form hydrogen bonds with the carboxyl group of IBU when it is adsorbed on the surface. During the release process, the solvent enters the IBU/matrix phase through the pores. The drug is then slowly dissolved into simulated body fluid (SBF) from the surface and diffuses from the system along the solvent-filled mesoporous channels. The loading amount of IBU in IBU–Gd2O3:Er3+@nSiO2@mSiO2 is 11 wt %. The cumulative drug release profile of IBU–Gd2O3:Er3+@nSiO2@mSiO2 system in SBF are depicted in Fig. 10A. It can be seen that the drug release rate is quite fast in the first hour and then become much slower. The initial release may be attributed to IBU being weakly adsorbed on the outer surface of mesoporous nanocomposite, and the slow release of the rest of IBU may be due to the strong interaction between IBU molecules and the surface.
 | ||
| Fig. 10 Cumulative IBU release from (A) the IBU–Gd2O3:Er3+@nSiO2@mSiO2 system versus release time in the release media of SBF. Up-conversion emission intensity of (B) Er3+ in IBU- Gd2O3:Er3+@nSiO2@mSiO2 as a function of the cumulatively released IBU. | ||
Here it is of great importance to investigate the relationship between the up-conversion emission intensity of the IBU–Gd2O3:Er3+@nSiO2@mSiO2 system and the cumulative release of IBU, as given in Fig. 10B. The up-conversion emission intensity of Er3+ increases with the cumulative release of IBU, reaching a maximum when the IBU is completely released from the drug storage system. It is well known that the emission of rare earth ions will be quenched to some extent in the environments that have a high phonon frequency.29 The organic groups in IBU, that have tremendous vibration frequencies from 1000 to 3500 cm−1 will quench the emission of Er3+ to a great extent in the IBU–Gd2O3:Er3+@nSiO2@mSiO2 system. With the release of IBU, the quenching effect will be weakened, which results in the increase of emission intensity. This correlation between the emission intensity and the extent of drug release is potentially a probe for monitoring the drug release efficiency during disease therapy.
4. Conclusion
In conclusion, Gd2O3:Er3+ nanoparticles have been successfully encapsulated by mesoporous silica using CTAB as template through a sol–gel process. The obtained core-shell structured composite material possesses bright up-conversion luminescence, and ordered hexagonal mesopores at room temperature. Our preliminary results showed that this bifunctional system is non-toxic, which can be potentially used as a drug delivery system, in which the drug release efficiency can be monitored by the up-conversion luminescence.
Acknowledgements
This project is financially supported by National Basic Research Program of China (2007CB935502, 2010CB327704), and the National Natural Science Foundation of China (NSFC 50702057, 50872131, 20901074, 20921002).References and Notes
- (a) K. T. Yong, I. Roy, M. T. Swihart and P. N. Prasad, J. Mater. Chem., 2009, 19, 4655 RSC; (b) J. H. Gao, H. W. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097 CrossRef CAS; (c) F. Wang, Y. Han, C. S. Lim, Y. H. Lu, J. Wang, J. Xu, H. Y. Chen, C. Zhang, M. H. Hong and X. G. Liu, Nature, 2010, 463, 1061 CrossRef CAS.
- (a) J. Kim, Y. Z. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372 RSC; (b) H. F. Yin, C. Wang, H. G. Zhu, S. H. Overbury, S. H. Sun and S. Dai, Chem. Commun., 2008, 4357 RSC; (c) J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421 CrossRef CAS; (d) C. C. Lin, Z. R. Xiao, G. Y. Guo, T. S. Chan and R. S. Liu, J. Am. Chem. Soc., 2010, 132, 3020 CrossRef CAS; (e) A. K. Sharma, K. H. Son, B. Y. Han and K. S. Sohn, Adv. Funct. Mater., 2010, 20, 1750 CAS; (f) L. D. Carlos, R. A. S. Ferreira, V. Z. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS.
- (a) L. Q. Xiong, Z. G. Chen, M. X. Yu, F. Y. Li, C. Liu and H. C. Huang, Biomaterials, 2009, 30, 5592 CrossRef CAS; (b) Z. Q. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765 CrossRef CAS; (c) S. Heer, O. Lehmann, M. Hasse and H. U. Güdel, Angew. Chem., Int. Ed., 2003, 42, 3179 CrossRef CAS; (d) G. S. Yi and G. M. Chow, J. Mater. Chem., 2005, 15, 4460 RSC; (e) Y. S. Liu, D. T. Tu, H. M. Zhu, R. F. Li, W. Q. Luo and X. Y. Chen, Adv. Mater., 2010, 22, 3266 CrossRef CAS; (f) J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417 RSC; (g) Q. Ju, W. Q. Luo, Y. S. Liu, H. M. Zhu, R. F. Li and X. Y. Chen, Nanoscale, 2010, 2, 1208 RSC.
- (a) F. Wang and X. G. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS; (b) O. Ehlert, R. Thomann, M. Darbandi and T. Nann, ACS Nano, 2008, 2, 120 CrossRef CAS; (c) R. Kumar, M. Nyk, T. Y. Ohulchanskyy, C. A. Flask and P. N. Prasad, Adv. Funct. Mater., 2009, 19, 853 CrossRef CAS; (d) J. W. Wang and P. A. Tanner, J. Am. Chem. Soc., 2010, 132, 947 CrossRef CAS; (e) P. Ghosh, A. Kar and A. Patra, Nanoscale, 2010, 2, 1196 RSC.
- (a) F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839 RSC; (b) L. Y. Wang, R. X. Yan, Z. Y. Hao, L. Wang, J. H. Zeng, J. Bao, X. Wang, Q. Peng and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 6054 CrossRef CAS; (c) S. Heer, K. Kompe, H. U. Gudel and M. Haase, Adv. Mater., 2005, 17, 2119 CrossRef CAS; (d) H. X. Mai, Y. W. Zhang, R. Si, Z. G. Yan, L. D. Sun, L. P. You and C. H. Yan, J. Am. Chem. Soc., 2006, 128, 6426 CrossRef CAS.
- (a) Z. Li, Y. Zhang, B. Shuter and N. M. Idris, Langmuir, 2009, 25, 12015 CrossRef CAS; (b) R. Sivakumar, F. C. J. M. van Veggel and M. Raudsepp, J. Am. Chem. Soc., 2005, 127, 12464 CrossRef CAS; (c) F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924 CrossRef CAS.
- (a) K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181 CrossRef CAS; (b) K. E. Fischer, B. J. Alemán, S. L. Tao, R. H. Daniels, E. M. Li and M. D. Bünger, Nano Lett., 2009, 9, 716 CrossRef CAS; (c) J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462 CrossRef CAS; (d) W. Wei, G. H. Ma, G. Hu, D. Yu, T. Mcleish and Z. G. Su, J. Am. Chem. Soc., 2008, 130, 15808 CrossRef CAS.
- (a) Q. Yang, S. C. Wang, P. W. Fan, L. F. Wang, Y. Di and K. F. Lin, Chem. Mater., 2005, 17, 5999 CrossRef CAS; (b) W. Zhao, H. Chen, Y. Li, L. Li, M. Lang and J. Shi, Adv. Funct. Mater., 2008, 18, 2780 CrossRef CAS; (c) A. Rösler, G. W. M. Vandermuelen and H. A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95 CrossRef CAS.
- (a) I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225 CrossRef CAS; (b) X. Guo and F. Szoka, Acc. Chem. Res., 2003, 36, 335 CrossRef CAS; (c) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS.
- M. Itokazu, W. Yang, T. Aoki, A. Ohara and N. Kato, Biomaterials, 1998, 19, 817 CrossRef CAS.
- M. A. Rauschmann, T. A. Wichelhaus, V. Stirnal, E. Dingeldein, L. Zichner, R. Schnettler and V. Alt, Biomaterials, 2005, 26, 2677 CrossRef CAS.
- R. P. Del Real, J. G. C. Wolke and M. Vallet-Regí, Biomaterials, 2002, 17, 3673 CrossRef.
- M. Bohner, Injury, 2000, 3, D37–D47 CrossRef.
- H. H. Yang, Q. Z. Zhu, H. Y. Qu, X. L. Chen, M. T. Din and J. G. Xu, Anal. Biochem., 2002, 308, 71 CrossRef CAS.
- P. Caliceti, S. Salmaso, A. Lante, M. Yoshida, R. Katakai and F. Martellini, J. Controlled Release, 2001, 75, 173 CrossRef CAS.
- M. Changez, K. Burugapalli, V. Koul and V. Choudhary, Biomaterials, 2003, 24, 527 CrossRef CAS.
- P. P. Yang, Z. W. Quan, Z. Y. Hou, C. X. Li, X. J. Kang, Z. Y. Cheng and J. Lin, Biomaterials, 2009, 30, 4786 CrossRef CAS.
- (a) Y. Lu, Y. Yin, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 183 CrossRef CAS; (b) O. M. Ntwaeaborwa and P. H. Holloway, Nanotechnology, 2005, 16, 865 CrossRef CAS; (c) Y. H. Deng, C. C. Wang, J. H. Hu, W. L. Yang and S. K. Fu, Colloids Surf., A, 2005, 26, 87 CrossRef.
- L. Levy, Y. Sahoo, K. S. Kim, E. J. Bergey and P. N. Prasad, Chem. Mater., 2002, 14, 3715 CrossRef CAS.
- X. Q. Xu, C. H. Deng, M. X. Gao, W. J. Yu, P. Y. Yang and X. M. Zhang, Adv. Mater., 2006, 18, 3289 CrossRef CAS.
- Y. Li, B. Yan, C. H. Deng, W. J. Yu, X. Q. Xu and P. Y. Yang, Proteomics, 2007, 7, 2330 CrossRef CAS.
- (a) C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS; (b) P. P. Yang, Z. W. Quan, L. L. Lu, S. S. Huang and J. Lin, Biomaterials, 2008, 29, 4341 CrossRef CAS; (c) M. Arruebo, M. Galán, N. Navascués, C. Téllez, C. Marquina and M. R. Ibarra, Chem. Mater., 2006, 18, 1911 CrossRef CAS.
- E. Matijević and W. P. Hsu, J. Colloid Interface Sci., 1987, 118, 506 CAS.
- M. Vallet-Regí, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308 CrossRef CAS.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- J. Yang, C. M. Zhang, C. Peng, C. X. Li, L. L. Wang, R. T. Chai and J. Lin, Chem.–Eur. J., 2009, 15, 4649 CrossRef CAS.
- M. Liong, S. Angelos, E. Choi, K. Patel, J. F. Stoddart and J. I. Zink, J. Mater. Chem., 2009, 19, 6251 RSC.
- G. Blasse, B. C. Grabmaier, Luminescent Materials, Springer, Berlin 1994, Ch. 4 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |