Synthesis and characterization of biomimetic hydroxyapatite/sepiolite nanocomposites†
Chaoying
Wan
and
Biqiong
Chen
*
Department of Mechanical and Manufacturing Engineering, Trinity Centre for Bioengineering, Trinity College Dublin, College Green, Dublin 2, Ireland. E-mail: chenb@tcd.ie; Fax: +353 1 679 5554; Tel: +353 1 896 1729
First published on 22nd November 2010
Abstract
Natural fibrous sepiolite with a high surface area, negative surface charge and porous structure is promising for hydroxyapatite (HAp) mineralization since the clay is naturally abundant and biocompatible. In this paper, the use of fibrous sepiolite as a template for growth of HAp nanocrystals was reported for the first time. Carbonated HAp nanorods with dimensions of 20–60 nm in length and 10–20 nm in diameter were successfully grown on the sepiolite surface with a preferred orientation to the c-axis. The critical nucleus radius of HAp in the presence of natural sepiolite was estimated as 0.296–0.312 nm. Strong acid-activation increased the specific surface area of the sepiolite by 205% and also transformed the sepiolite to silica fiber with an elastic modulus being 395% of the original value. The novel HAp/acid-activated sepiolite biocomposite has a specific surface area of 182 m2 g−1 and an elastic modulus of over 20 GPa, considerably higher than those of the HAp synthesized without sepiolite. Such hierarchically assembled HAp/sepiolite biocomposites with controlled size and improved modulus open a new way to expand the applications of naturally abundant clays in biological load-bearing devices.
Introduction
Nanosized crystalline calcium phosphates in the form of apatites naturally occur as the basic inorganic contents of bones and teeth of mammals as a result of natural selection.1 Due to their large specific surface area, these apatites have strong interactions with proteins which are the major organic constituents in bones.2 The biological hydroxyapatite (Ca10(PO4)6(OH)2, HAp) exists as homogeneous plate-like nanocrystals with the size of 15–30 nm wide and 30–50 nm long in bones, and as rod-like nanocrystals of 25–100 nm thick with lengths in the range of 100 nm to several microns in enamel.3 Taking the inspiration from the natural tissues, synthesis of HAp nanocrystals with well-controlled sizes and shapes is regarded as an important prerequisite for biomineralization and biomimetic assembly of biocomposites.1,4a Compared to their microscaled counterparts, nanosized hydroxyapatites exhibit improved sinterability and densification, better bioactivity, superior biocompatibility and osteointegration of bone graft substitutes,4 and hence much enhanced performances because of their larger surface-to-volume ratios and unusual chemical synergistic effects.The particle size and crystallinity of HAp are generally influenced by the presence of organic modifiers and reaction conditions.4 For example, spherical HAp nanoparticles with diameters of 20 ± 5 nm, 40 ± 10 nm and 80 ± 12 nm were formed by varying the surfactant (hexadecyltrimethylammonium bromide) concentration in the range of 6.0 × 10−4 to 12.0 × 10−4 M.4bHAp nanorods with dimensions from Φ 12.4 nm × 30.0 nm to Φ 17.9 nm × 48.7 nm were obtained by using different types of surfactants, and the crystallinity increased with the increasing synthesis temperatures.5 Microemulsion, micelle and reverse micelles, polymer capsules, Langmuir–Blodgett films and self-assembled monolayers1,4b,c provide confined environment for synthesis of HAp nanocrystals, in which the morphology and size of HAp were mainly dependent on the ‘nanoreactor’ profile and reaction condition.
Nanoparticles, such as titanium nanotubes,6carbon nanotubes,7 mesoporous silica8 and gold nanoparticles,9 have also been used as templates for mineralization of nanocrystalline HAp, in which their dimensions and physiochemical properties play a key role in regulation of HAp growth.6–9 The nucleation activity of HAp was enhanced when synthesized with nanostructured titanium nanotubes.6aCarboxyl-functionalized multi-walled carbon nanotubes (MWNTs)7c and PEGylation of MWNTs7d have been used as matrices for HAp growth based on the potential of carboxylate ions to bind Ca2+ ions onto their surfaces. Silica bioceramics are capable of promoting apatite precipitation and have been used as prosthetic bone and dental implants.8b Porous bioglass ceramics induce nucleation of HAp nanocrystals inside the pores where the pore size is a main factor governing the nanocrystal growth. It was found that bone bonding occurred when the bioglass porosity resulted in surface areas greater than 80 m2 g−1,8c that pore sizes less than 2 nm were not suitable for inducing HAp growth,8d and that the critical nucleation diameter of the HAp on CaO–SiO2 substrate was above 2.7 nm but below 4.9 nm.8e Macroporous CaO–4SiO2 bioglass with a pore diameter of about 400 nm could form a full layer of HAp covering the pores when it was soaked in simulated body fluid at 37 °C.8f As a summary of the above research, negatively charged and porous substrates are favorable for HAp mineralization.
As an alternative nanoparticle template, natural fibrous sepiolite nanoclay, (Si12)(Mg8)O30(OH)4(OH2)48·H2O, with periodic structural intervals along the b-axis and micropore tunnels of dimensions of 0.37 × 1.06 nm2 (ref. 10b) along the c-axis (Fig. 1a and b) providing a high specific surface area up to 320 m2 g−1, a porous volume of 0.4 cm3 g−1,10a and a negative surface charge may accomplish the structural requirements for growing HAp nanocrystals with controlled sizes. Meanwhile, the roles of the nanometric tunnels and pore defects (2–20 nm in diameter)10c in sepiolite structures in the HAp mineralization are highly in need of elucidation. Furthermore, acid-activated sepiolites owning higher porosity due to the acid-extraction of magnesium ions from the octahedral sheets10d and abundant silanol groups on the surface10e make them more promising to induce HAp biomineralization.
 | ||
| Fig. 1 TEM images illustrating the structures of the sepiolites before and after acid-activation: (a) scheme of sepiolite structure and dimensions; (b) the pristine sepiolite; (c) the high magnification image of the sepiolite showing 1.22 nm lattice-fringe separations; and (d) Acid-SEP2. | ||
Clay minerals have widely been applied in pharmaceutical and biological applications due to their excellent properties such as high absorption/desorption capacity, ion exchange capacity, rheology, low or no toxicity and good biocompatibility.11 They have been proven to be effective inorganic hosts for proteins, drugs, bioactive molecules, and magnetic nanoparticles,12,13etc. To the authors' knowledge, the use of clay for bioactive HAp mineralization has not been reported. In this paper, fibrous sepiolite was used as a template for in situgrowth of HAp. The effects of physiochemical properties of the pristine sepiolite and two acid-activated sepiolites on the nucleation and morphology of HAp crystals were investigated. The resultant HAp/sepiolite biocomposites are expected to give better mechanical properties and similar bioactivity compared to HAp considering the high elastic modulus of acid-activated sepiolites and synergistic effects, which have great potential to be applied in polymer–inorganic tissue scaffolds that often suffer the problems with low strength and modulus.
Experimental
Materials
A high purity sepiolite (Pansil S1500) supplied by Tolsa UK Ltd was used as received. The particle size was smaller than 5 µm as determined by Field Emission Scanning Electron Microscopy. Hydrochloric acid (HCl), calcium nitrate (Ca(NO3)2·4H2O) and diammonium hydrogen phosphate ((NH4)2HPO4) were purchased from Sigma-Aldrich.Surface-modification of sepiolite
Five grams of sepiolite clay were treated in 150 ml of 1 M HCl at 100 °C under rigorous stirring for either 30 min or 3 h to produce different acid-activated sepiolites denoted as Acid-SEP1 and Acid-SEP2. Then, the acid-treated sepiolites were centrifuged and washed with distilled water for several times until the pH value of the final solution reached 7. Finally they were oven-dried at 100 °C for 12 h and ground into fine powders.Mineralization using sepiolites
HAp was synthesized by using the pristine sepiolite or Acid-SEP(1,2) as a substrate, and the amounts of calcium and phosphate ions added for HAp formation were determined to give the final weight ratio of HAp to the substrate of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The aqueous sepiolite suspension of 1% (w/v) was prepared and stirred at 50 °C overnight. The calcium ions solution (0.5 M) and phosphate ions solution (0.3 M) were prepared separately and pre-adjusted to pH = 10 by use of an ammonium solution. The Ca2+ solution was added to the sepiolite suspension and kept stirring for 2 h, which was followed by a dropwise addition of PO43− ions to give the stoichiometric Ca2+/PO43− ratio of 1.67 for HAp formation. The pH was maintained at 10 throughout the experiment. The resultant suspension was stirred at 50 °C for 24 h and aged for 48 h at room temperature. The samples named as HAp/SEP, HAp/Acid-SEP1, and HAp/Acid-SEP2 were washed repeatedly with distilled water and ethanol before freeze-drying. HAp in the absence of the sepiolites was prepared in parallel for comparison.
:1. The aqueous sepiolite suspension of 1% (w/v) was prepared and stirred at 50 °C overnight. The calcium ions solution (0.5 M) and phosphate ions solution (0.3 M) were prepared separately and pre-adjusted to pH = 10 by use of an ammonium solution. The Ca2+ solution was added to the sepiolite suspension and kept stirring for 2 h, which was followed by a dropwise addition of PO43− ions to give the stoichiometric Ca2+/PO43− ratio of 1.67 for HAp formation. The pH was maintained at 10 throughout the experiment. The resultant suspension was stirred at 50 °C for 24 h and aged for 48 h at room temperature. The samples named as HAp/SEP, HAp/Acid-SEP1, and HAp/Acid-SEP2 were washed repeatedly with distilled water and ethanol before freeze-drying. HAp in the absence of the sepiolites was prepared in parallel for comparison.
Characterization
Transmission Electron Microscopy (TEM) images were recorded on a JEOL 2011 microscope operated at 200 kV. The freeze-dried particles were dispersed in ethanol and then deposited on carbon-coated copper grids. X-Ray Diffraction (XRD) measurements were performed on a Phillips PW1720 power X-ray diffractometer equipped with a multichannel detector by use of a Cu Kα1 (λ = 0.15406 nm) monochromatic X-ray beam. All the samples were measured within 2θ of 2–70° with a scan rate of 1° per min. The average crystallite size (Dp) of HAp was calculated based on Scherrer's equation14a using the (002) diffraction peak from the XRD pattern. The crystallinity (Xc) was calculated according to β002(Xc)1/3 = KA,14b where β002 is in degree and KA is a constant, i.e. 0.24. X-Ray fluorescence (XRF) quantitative measurements were carried out using a sequential Siemens SRS3000 spectrometer with a Rh-anode X-ray tube (75 µm Be window), a 60 kV generator and an eight-position crystal changer. Thermogravimetric analyses (TGA) were performed on a Perkin Elmer Pyrus 1 TGA under nitrogen atmosphere. The temperature range was from 30 to 900 °C with the heating rate of 10 °C min−1. The FT-IR spectra were recorded on a Perkin Elmer Spectrum One NTS analyzer (resolution 4 cm−1) at room temperature. The specific surface areas were measured by N2 adsorption–desorption isotherms using a fully automated micromeritics Tristar 3000 BET surface area analyzer. Prior to the analysis, the moisture adsorbed on the surface of the fine powders was eliminated by preheating the powders at 150 °C for 2 h, and the samples were degassed at 250 °C for 3 h. Densities were determined by helium pycnometry using a MicroMeritics AccuPyc 1330 Pycnometer.Nanoindentation experiments were carried out using an MTS nanoindenter XP (MTS Systems) with a load-control method. The indenter system was calibrated using a fused silica standard sample (measured modulus of 73 GPa). A Berkovich diamond indenter with a total included angle of 142.3° and 50 nm nominal radius was used throughout the measurement. Indentation tests were conducted at a loading/unloading rate of 0.05 mN s−1, with a hold time of 30 s at a maximum load of 50 mN to minimize the influence of creep on unloading. A drift rate of 0.05 nm s−1 was applied to account for thermal expansion or contraction occurred in the test specimen and/or the indenter. Load-displacement curves were recorded and ten indents were made on each sample. The samples were uniaxially pressed at 8 tons to form disks with 13 mm diameter and 3 mm thick dimensions, and then calcinated in a furnace at 1200 °C for 2 h with a heating rate of 10 °C min−1. The elastic modulus and hardness were determined using the Oliver–Pharr method,15 as detailed below.
The hardness H was calculated by dividing the maximum applied load, Pmax, by the contact area, A, that is,15a
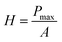 | (1) |
The elastic modulus E of the samples is obtained by the following equation:
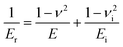 | (2) |
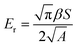 | (3) |
Results and discussion
Characterization of the pristine and acid-treated sepiolites
FT-IR spectra of the pristine sepiolite and acid-activated sepiolites are shown in Fig. 2. In Fig. 2A (curve a), the absorption bands at 3762–3560 cm−1 are ascribed to the OH stretching vibration from Mg3OH units in the octahedral structure of sepiolite.17a Broad bands near 3370 and 3240 cm−1 are due to H–O–H vibrations of adsorbed water. The absorption bands in the range of 1300 and 500 cm−1 are mainly ascribed to Si–O bonds in the tetrahedral sheet. The wide band centered at 974 cm−1 is due to Si–O–Si in-plane vibration. The absorption at 787 cm−1 is due to OH deformation vibration,17b and the bands at around 694 and 644 cm−1 are ascribed to the Mg3OH bending vibrations.17c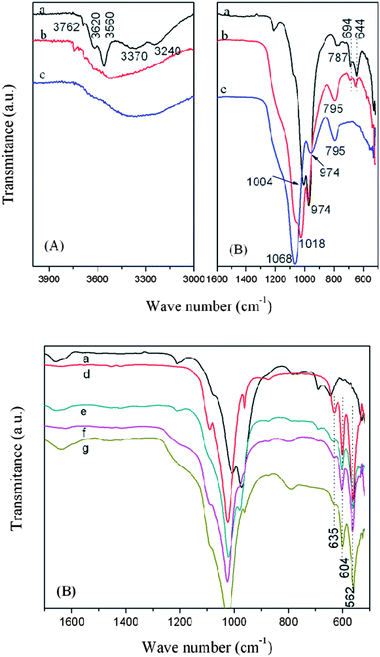 | ||
| Fig. 2 FT-IR absorption spectra of the pristine sepiolite (a), Acid-SEP1 (b) and Acid-SEP2 (c) in the OH stretch and 1600–500 cm−1 regions (A); the spectra of the pristine sepiolite (a), HAp (d), HAp/SEP (e), HAp/Acid-SEP1 (f), and HAp/Acid-SEP2 (g) biocomposites in 1600–500 cm−1 region (B). | ||
For the Acid-SEP1 (Fig. 2A, curve b), the absorption bands at 3560–3620 cm−1 ascribed to the Mg–OH groups disappeared but the band near 3762 cm−1 still existed. The Si–O absorption bands including 1211, 1078, 1006, and 974 cm−1 in sepiolite changed their shapes, shifted or disappeared, and the bands at around 694 and 644 cm−1 with lower intensities were still observed in Fig. 2A. This indicates that part of the Mg–OH bonds in the sepiolite was ruptured due to the acid attack.17d
For the Acid-SEP2 sample (Fig. 2A, curve c), the characteristic absorption bands for Mg–OH at around 3762, 694 and 644 cm−1 disappeared indicating the absence of Mg–OH in the octahedral sheet of Acid-SEP2 sample. The bands at 1057 and 1018 cm−1 combined into one peak and shifted to 1068 cm−1 which is associated to amorphous silica.17e Another characteristic band of amorphous silica near 795 cm−1 increased in intensity18a as the progress of acid dissolution. The changes of the Si–O stretching band and the gradual movement to 1068 cm−1 indicate successive release of the Mg atoms from the octahedra as well as removal of Mg from the tetrahedral sheets.18b
Fig. 3A shows the XRD patterns of the pristine and acid-treated sepiolites. The pristine sepiolite (curve a) displays an intense peak at 2θ = 7.27° (d-spacing of 1.22 nm), corresponding to the internal channel reflections,19 which is also observed by TEM (Fig. 1c). The diffraction peaks of sepiolite decreased in intensity during the mild acid-treatment as observed in Acid-SEP1 (curve b). The disappearance of the characteristic peaks, especially the sharp peak at 2θ = 7.27°, and appearance of a broad peak in the range of 15° to 30° associated to amorphous silica indicate that the crystalline structure of sepiolite collapses during the strong acid treatment.10e Along with the FT-IR characterization as discussed above and the XRF results presented in Table S1†, this confirms that the magnesia layers have been completely removed from the sepiolite crystalline lattice and the sepiolite has been converted to silica after the strong acid treatment. As a result, the typical fiber diameter of the sepiolite is reduced from ∼30 to ∼5 nm, the aspect ratio is increased from ∼20 to 300 (Fig. 1b and d), and the specific surface area is increased from 148.2 to 452.5 m2 g−1 (Table 1). The increase of the specific surface area by 205% is in excellent agreement with the structural change experienced by the clay during the strong acid-treatment in which case a crystalline lattice consisting of two layers of silica and one layer of magnesia has become two crystalline lattices each consisting of one layer of silica only: the original lattice has a mass equivalent to about 3 times of the new single lattice yet gives the same surface area by neglecting the side surfaces because of the high aspect ratio of the lattice. The BJH pore volume and pore diameter were also found to increase by 59% and 99% (Table 1), respectively. Such acid-activated sepiolites with porous structure, high aspect ratio and high density of SiOH groups are expected to be more favorable for nucleation and growth of HAp crystallites as opposed to the pristine sepiolite.
| Sample | S BET/m2 g−1 | BJH pore volume/cm3 g−1 | BJH pore diameter/nm |
|---|---|---|---|
| Sepiolite | 148.2 | 0.5722 | 7.9 |
| Acid-SEP2 | 452.5 | 0.9076 | 15.7 |
| HAp | 109.3 | 0.4272 | 3.4 |
| HAp/SEP | 145.7 | 0.6011 | 10.9 |
| HAp/Acid-SEP2 | 182.0 | 0.7916 | 11.1 |
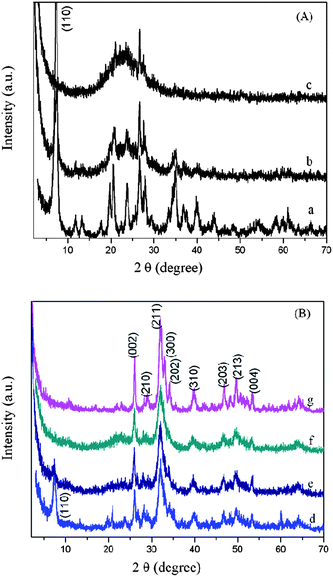 | ||
| Fig. 3 XRD patterns of (A) the pristine sepiolite (a), Acid-SEP1 (b) and Acid-SEP2 (c); (B) HAp/SEP (d), HAp/Acid-SEP1 (e), HAp/Acid-SEP2 (f), and HAp (g). | ||
Characterization of nanocrystalline hydroxyapatites grown on the sepiolites
In Fig. 2B (curve d), the HAp exhibits absorption bands at 562, 604, 962, 1024 and 1090 cm−1 which are the characteristic peaks of PO43−. The absorption band at 635 cm−1 belongs to librational bands of hydroxyl groups in HAp. The bands at 1416 cm−1 and 873 cm−1 are associated to the C–O stretching vibration mode on the CO32−groups, which might be due to the dissolved CO2 from atmosphere during the reactions.20 The HAp/SEP and HAp/Acid-SEP(1,2) samples exhibit absorption bands at 604 and 562 cm−1 corresponding to the O–P–O bending vibrations which are considered to originate from the PO43−groups in HAp crystals. The bands at 1094, 1025 and 961 cm−1 designated to P–O stretching vibration modes partially overlapped with those for Si–O groups because of the similarities of the SiO44− and PO43− tetrahedral units.20 As characterized by XRD (curve g in Fig. 3B), diffraction peaks at 2θ of 26.1°, 32.2°, 53.9° and 64.7° corresponding to HAp (JCPDS 09-0432) were observed, and no obvious traces of brushite (especially at 2θ = 12° and 21°) were identified, which means the synthesized HAp is pure.21 The characteristic diffraction peaks of HAp are visible for the HAp/sepiolite samples. The above FT-IR and XRD results indicate that HAp has successfully grown on both pristine sepiolite and acid-activated sepiolites.The average crystallite size (Dp) and crystallinity (Xc) of HAp/SEP biocomposite calculated from the XRD traces, being 21.1 nm and 20.9%, respectively, are lower than those of HAp being 27.6 nm and 47.2% (Table 2). Low-crystallinity apatites are preferred in biomedical and tissue engineering applications due to their high ‘in vivo’ resorbability rate.22 The Dp of HAp/SEP does not show much difference as compared to those of HAp/Acid-SEP samples, but its Xc is slightly lower, which may be attributed to the more Si–OH groups present in the latter acting as the nucleation sites. The Dp and Xc values of the HAp/sepiolite biocomposites are comparable to those of HAp nanocrystals synthesized by using chitosan as the template21 and those of biological apatites.22
| Sample | Line width (002) FWHM/° | Average crystallite size (002), Dp/nm | Crystallinity, Xc (%) |
|---|---|---|---|
| HAp | 0.308 | 27.6 | 47.2 |
| HAp/SEP | 0.405 | 21.1 | 20.9 |
| HAp/Acid-SEP1 | 0.391 | 21.8 | 23.1 |
| HAp/Acid-SEP2 | 0.375 | 22.7 | 26.3 |
The BET specific surface areas of the HAp/SEP and HAp/Acid-SEP2 are considerably higher than that of the HAp nanocrystals synthesized without sepiolite, being 145.7 m2 g−1 and 182.0 m2 g−1versus 109.3 m2 g−1 (Table 1). These suggest the HA/sepiolite composites would provide larger interfacial surfaces for manipulating their interactions with surroundings when compared to the unfilled HAp. As observed with TEM, the HAp nanocrystals are needle-like of ca. 10 × 100 nm in size (Fig. 4c).
 | ||
| Fig. 4 TEM images illustrating HAp formation on the sepiolites: (a) HAp/SEP biocomposite, the circled part shows the broken sepiolite surface (defects) also acts as nucleation sites for HAp nanoparticles growth; (b) high magnification image of the HAp nanocrystals showing 0.35 nm lattice-fringe separations in the HAp/SEP sample, the arrow indicates the distance between the two HAp crystals on the sepiolite surface, which is 6.25 nm in this case; (c) HAp particles synthesized in the same condition as that of HAp/SEP sample; (d) HAp/Acid-SEP2 biocomposite. | ||
In comparison, the morphology of HAp in HAp/SEP biocomposite (Fig. 4a) is in nanorod-shape with 20–60 nm in length and 10–20 nm in width. The nanorods grew from the external channels and oriented to the c-axis of sepiolite fibers, with some growing along the broken surfaces (defects). A HAp lattice plane of 0.35 nm corresponding to the (002) plane was clearly identified in the TEM image (Fig. 4b). A 6.25 nm-wide zone with a different crystalline structure in between the two HAp nanocrystals was also observed. The amount of HAp on the sepiolite surface was estimated as 31 wt% based on the TGA results by comparing the weight losses of HAp, sepiolite and HAp/SEP composite in the range of 300–600 °C (Fig. S1†). As shown in Fig. 4d, more HAp crystals grew inside Acid-SEP2 textures and interconnected closely with the silica fibers, indicating that the size and amount of HAp nanocrystals directly correlate with the amount of Si–OH groups (nucleation sites) in sepiolites. The amount of HAp crystals on the Acid-SEP2 structures was estimated as 67 wt% based on the TGA results, similar to the value estimated from the XRF result by considering the contents of CaO in Acid-SEP2, HAp and HAp/Acid-SEP2 (Table S1†), being 65 wt%. So, the average ratio of HAp to the Acid-SEP2 (1.94:1) is around the initial feed ratio (2:1) indicating the nucleation and growth of HAp crystals mainly occurred on the Acid-SEP2 fibers. The estimated amount of HAp in the Acid-SEP1 is 48 wt%, which is in between the values for HAp/SEP and HAp/Acid-SEP2 biocomposites. Therefore, the acid-activated sepiolite is more favorable for the nucleation and growth of HAp nanocrystals due to the presence of more Si–OH functional groups acting as nucleation sites, a larger specific surface and more mesopores acting as growth templates.
Based on the above analysis, the nucleation mechanism of HAp on sepiolite and acid-activated sepiolites is proposed as follows. The initial Si–OH groups on the edge of surface channels and some pore defects in the pristine sepiolite may absorb Ca2+ ions through electrostatic interactions and act as nucleation sites for HAp formation. In comparison, the abundant Si–OH groups on the acid-activated sepiolite surface, i.e. the freshly exfoliated silicate fibers, are expected to attract even more Ca2+ ions into the mesopores of sepiolites through an inter-diffusion process. Moreover, some Ca2+ ions could enter into the octahedral positions originally occupied by Mg2+ which were lixiviated under strong acid attack, thus creating local supersaturation of Ca2+ ions in the channels and around the silica fibers, and facilitate nucleation and growth of HAp nanocrystals on the sepiolite templates.
Nucleation of crystalline hydroxyapatites in the presence of sepiolite
According to the classical nucleation theory, the driving force for a small nucleus to overcome a nucleation barrier and become a stable growing crystal in the solution is the change in Gibbs free energy. For a homogenous nucleation process, the total Gibbs free energy ΔGhomo for the formation of a spherical crystallite with radius r is expressed as eqn (4),23,24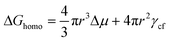 | (4) |
In the presence of sepiolite, nucleation of HAp crystallites takes place in a heterogeneous way, and the nucleation barrier is reduced to,24a
| ΔGhet = f(m,R′)ΔGhomo [0 ≤ f(m,R′) ≤ 1] | (5) |
Assuming the sepiolite is a flat substrate, Rs → ∞, R′ → ∞, so f(m,R′) = f (m) which is only dependent on m,24a thus:
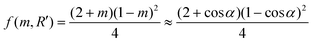 | (6) |
The interfacial tension γij between phases i and j is obtained using the van Oss–Chaudhury–Good equation,23a
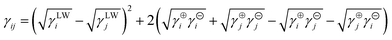 | (7) |
According to the surface tension components cited from the literature as shown in Table 3, the γsf, γcf and γsc were calculated as 9.75, 9.06, and 0.17 mJ m−2, respectively. Then m → 1, α ≈ 0°, f (m) → 0, which means both a strong interaction and an optimal structural match occur between the substrate (sepiolite) and the nucleating phase (HAp); the heterogeneous nucleation is kinetically favored.23a,24 Nucleation of HAp crystallites tends to be templated by sepiolite having an excellent structural correlation with the crystalline phase of HAp.
The heterogeneous critical nucleus size rc′ is obtained by evaluating from (∂ΔGhet/∂r) = 0, so
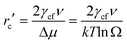 | (8) |
From eqn (8), the critical nucleus size of rc′ is dependent on the interfacial tension and supersaturation. The interfacial tension of HAp, γcf, has been determined by different methods including crystal growth and dissolution kinetics as well as contact angle measurements as reviewed by Wu and Nancollas.23a The γcf values determined by the kinetics method vary from 9.3–120 mJ m−2 in the literature because the kinetics method involves a number of variables that are challenging to measure experimentally.23a In comparison, the contact angle measurement gives more consistent results and 10 mJ m−2 was determined for the γcf by contact angle methods.23a In our case, the γcf value was calculated as 9.06 mJ m−2 as previously discussed, which is in the same order of the measured data, 10 mJ m−2. Based on the estimated ln Ω values, eqn (8) and the data in Table 3, the rc′ is estimated to be 0.296–0.312 nm, thus the molecular volume of the unit cell of crystal is 1.867–2.187 nm3 given that nine molecules form one HAp unit cell.
The rc′ estimated in this study is in the same order of the reported data, varying from 0.28 to 0.72 nm.25–27 It is difficult to calculate the exact size and distribution of the tremendous numbers of nuclei in the presence of sepiolites since the nature and size of the nuclei change rapidly which induces the change of interfacial energy and residual supersaturation when precipitation occurs. However, the values of rc′ calculated based on the data in the literature would reflect the crystallization nature of HAp in the presence of sepiolites. As aforementioned, the acid-activated sepiolites provide more nucleation sites for HAp growth compared to the pristine sepiolite, so the size of critical nuclei of HAp in the presence of acid-activated sepiolites is speculated to be smaller than in the case with the pristine sepiolite.
Mechanical properties of calcinated HAp/sepiolite biocomposites
Elastic modulus and hardness of sepiolites and HAp calcinated at 1200 °C were measured by nanoindentation. The typical load-displacement curves and the results are shown in Fig. S2† and Table 4, respectively.| Sample code | Elastic modulusa/GPa | Hardnessa/GPa | Specific gravityb/g cm−3 |
|---|---|---|---|
| a The elastic moduli and hardness of the samples were measured by a nanoindenter and calculated with Oliver–Pharr method.15a b The specific gravity values were measured by helium pycnometry. | |||
| Sepiolite | 10.58 ± 0.22 | 1.31 ± 0.04 | 2.38 |
| Acid-SEP1 | 35.31 ± 2.19 | 3.75 ± 0.02 | 2.09 |
| Acid-SEP2 | 41.83 ± 3.51 | 5.91 ± 0.01 | 1.98 |
| HAp | 19.33 ± 0.05 | 1.69 ± 0.02 | 2.92 |
The elastic modulus of sepiolite is 10.58 GPa, much lower than the theoretical elastic modulus of smectite clay platelets without gallery spacing, namely about 230 GPa,16b due to the structural tunnels and pore defects present throughout the sepiolite textures. Vanorio et al.28a found the Young's modulus values for bulk montmorillonite and other smectite clay minerals were between 9.8 and 15.4 GPa by extrapolation of acoustic measurements on cold-pressed samples having different porosities, which are similar to those obtained with atomic force acoustic microscopy for dickite by Prasad et al.,28bi.e. 6.2 GPa. The elastic modulus of sepiolite measured by nanoindentation in this study is comparable to the literature data for bulk clays with gallery spacing. As presented in Table 4, the elastic modulus values of acid-activated sepiolites were higher than that of sepiolite, especially for the Acid-SEP2 with a 295% increase. This may be mainly explained as that the lixiviation of magnesium layers during the acid treatment has caused the sepiolite fibers to transform into silica fibers. However, the elastic modulus and density of Acid-SEP2, i.e. 41.83 GPa and 1.98 g cm−3, are lower than those of fused silica, 73 GPa and 2.2 g cm−3,15c respectively, suggesting the resultant silica fibers still have some structural defects (pores) and channels on the surface. Similar to modulus, the hardness values of acid-activated sepiolites were also higher than that of sepiolite. The elastic modulus and hardness of HAp were 19.33 GPa and 1.69 GPa, respectively, lower than those of the acid-activated sepiolites, while the density is substantially higher (Table 4, column 3).
Based on the experimental results given in Table 4 and the assumption that the HAp nanocrystals only grow on the fiber surfaces, the elastic modulus for the HAp/Acid-SEP2 biocomposite was predicted using Halpin–Tsai model,29
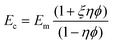 | (9) |
The calculated elastic modulus of HAp/Acid-SEP2 biocomposite is 29.03 GPa, 50% higher than that of HAp. The actual modulus of such composite is expected to be even higher because the HAp nanocrystals grow interconnecting with the fibers, existing both in the pore defects and the surface of the fibers as discussed above.
Conclusions
The natural fibrous sepiolite has been proven to be an effective inorganic host for HAp mineralization. Carbonated HAp nanorods with dimensions of 20–60 nm in length and 10–20 nm in width were observed to grow on the sepiolite surfaces with a preferred orientation to the c-axis. The critical nucleus radius of HAp in the presence of natural sepiolite was estimated as 0.296–0.312 nm which provides the evidence that the nucleation of HAp only occurs from the external channels and pore defects in the sepiolite, but not from the structural tunnels. Strong acid-activation increased the specific surface area of the sepiolite by 205%, and provided many more silanol groups on the fiber surfaces, facilitating formation of uniform and thick layers of HAp nanocrystallites on the surfaces. The novel HAp/Acid-SEP2 biocomposite developed has a specific surface area of 182 m2 g−1, an elastic modulus of over 20 GPa, and similar biological functions to HAp because of the full coverage of HAp nanocrystallites on the silica surfaces. This class of new biocomposites is considered to be promising in tissue engineering and medical fields.Acknowledgements
This research was supported by a Marie Curie International Incoming Fellowship within the 7th European Community Framework Programme under Grant Agreement No. PIIF-GA-2009-236739. C.W. also thanks funding from National Natural Science Foundation of China (Project No. 50803035).References
- (a) C. Sanchez, H. Arribart and M. M. G. Guille, Nat. Mater., 2005, 4, 277 CrossRef CAS; (b) S. Mann, in Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- R. J. Narayan, P. N. Kumta, C. Sfeir, D. H. Lee, D. Choi and D. Olton, JOM, 2004, 56, 38 CrossRef CAS.
- (a) S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271 CrossRef; (b) J. D. Currey, Science, 2005, 309, 253 CrossRef CAS.
- (a) Y. Cai and R. K. Tang, J. Mater. Chem., 2008, 18, 3775 RSC; (b) Y. Cai, Y. K. Liu, W. Q. Yan, Q. H. Hu, J. H. Tao, M. Zhang, Z. L. Shi and R. K. Tang, J. Mater. Chem., 2007, 17, 3780 RSC; (c) S. Bose and S. K. Saha, Chem. Mater., 2003, 15, 4464 CrossRef CAS; (d) T. J. Webster, C. Ergun, R. H. Doremus, R. W. Siegel and R. Bizios, Biomaterials, 2001, 22, 1327 CrossRef CAS; (e) E. S. Thian, J. Huang, S. M. Best, Z. H. Barber, R. A. Brooks, N. Rushton and W. Bonfield, Biomaterials, 2006, 27, 2692 CrossRef CAS.
- A. L. Wang, D. Liu, H. B. Yin, H. X. Wu, Y. J. Wada, M. Ren, T. S. Jiang, X. N. Cheng and Y. Q. Xu, Mater. Sci. Eng., C, 2007, 27, 865 CrossRef CAS.
- (a) S. H. Oh, R. R. Finones, C. Daraio, L. H. Chen and S. H. Jin, Biomaterials, 2005, 26, 4938 CrossRef CAS; (b) H. Tsuchiya, J. M. Macak, L. Müller, J. Kunze, F. Müller, P. Greil, S. Virtanen and P. Schmuki, J. Biomed. Mater. Res., Part A, 2006, 77, 534 CrossRef.
- (a) B. Zhao, H. Hu, S. K. Mandal and R. C. Haddon, Chem. Mater., 2005, 17, 3235 CrossRef CAS; (b) S. Liao, G. F. Xu, W. Wang, F. Watari, F. Z. Cui, S. Ramakrishna and C. K. Chan, Acta Biomater., 2007, 3, 669 CrossRef CAS; (c) S. Aryal, K. C. Remant Bahadur, N. Dharmaraj, K. W. Kim and H. Y. Kim, Scr. Mater., 2006, 54, 131 CrossRef CAS; (d) Y. Xiao, T. Gong and S. B. Zhou, Biomaterials, 2010, 31, 5182 CrossRef.
- (a) M. Iafisco, M. Marchetti, J. G. Morales, M. A. Hernandez-Hernandez, J. M. G. Ruiz and N. Roveri, Cryst. Growth Des., 2009, 9, 4912 CrossRef CAS; (b) N. Sahai and J. A. Tossell, J. Phys. Chem. B, 2000, 104, 4322 CrossRef CAS; (c) M. M. Walker, MS thesis, University of Florida, 1977; (d) M. M. Pereira and L. L. Hench, J. Sol-Gel Sci. Technol., 1996, 7, 59 CAS; (e) Y. Deng, X. K. Li and Q. Li, Ind. Eng. Chem. Res., 2009, 48, 8829 CrossRef CAS; (f) H. Yan, K. Zhang, C. F. Blanford, L. F. Francis and A. Stein, Chem. Mater., 2001, 13, 1374 CrossRef CAS.
- D. Rautaray, S. Mandal and M. Sastry, Langmuir, 2005, 21, 5185 CrossRef CAS.
- (a) P. Aranda, R. Kun, M. A. Martín-Luengo, S. Letaïef, I. Dékány and E. Ruiz-Hitzky, Chem. Mater., 2008, 20, 84 CrossRef CAS; (b) J. Santaren, J. Sanz and E. Ruiz-Hitzky, Clays Clay Miner., 1990, 38, 63 CrossRef CAS; (c) M. Rautureau and A. Mifsud, Clay Miner., 1977, 12, 309 CrossRef CAS; (d) J. L. Ahlrichs, J. C. Serna and J. M. Serratosa, Clays Clay Miner., 1975, 23, 119 CrossRef CAS; (e) S. Balci, J. Chem. Technol. Biotechnol., 1996, 66, 72 CrossRef CAS.
- J. H. Choy, S. J. Choi, J. M. Oh and T. Park, Appl. Clay Sci., 2007, 36, 122 CrossRef.
- V. Vergaro, E. Abdullayev, Y. M. Lvov, A. Zeitoun, R. Cingolani, R. Rinaldi and S. Leporatti, Biomacromolecules, 2010, 11, 820 CrossRef CAS.
- V. P. Cerezo, R. Sanchez, I. Salcedo and C. Aguzzi, Appl. Clay Sci., 2010, 48, 291 CrossRef.
- (a) B. D. Culity, in Elements of X-Ray Diffraction, Addison-Wesley, Reading, MA, 1978 Search PubMed; (b) E. Landi, A. Tampieri, G. Celotti and S. Sprio, J. Eur. Ceram. Soc., 2000, 20, 2377 CrossRef CAS.
- (a) W. C. Oliver and G. M. Pharr, J. Mater. Res., 2004, 19, 3 CrossRef CAS; (b) N. A. Sakharova, J. V. Fernandes, J. M. Antunes and M. C. Oliveira, Int. J. Solids Struct., 2009, 46, 1095 CrossRef; (c) C. P. Wong and R. S. Bollampally, J. Appl. Polym. Sci., 1999, 74, 3396 CrossRef CAS.
- (a) B. Chen and J. R. G. Evans, Macromolecules, 2006, 39, 747 CrossRef CAS; (b) B. Chen and J. R. G. Evans, Scr. Mater., 2006, 54, 1581 CrossRef CAS; (c) M. Akao, H. Aoki and K. Kato, J. Mater. Sci., 1981, 16, 809 CrossRef CAS.
- (a) J. D. Russell, Infrared Methods, in Handbook of Determinative Methods in Clay Mineralogy, ed. M. J. Wilson, Chapman & Hall, New York, 1987 Search PubMed; (b) R. L. Frost, O. B. Locos, H. Ruan and J. T. Kloprogge, Vib. Spectrosc., 2001, 27, 1 CrossRef CAS; (c) J. Cornejo and M. C. Hermosin, Clay Miner., 1988, 23, 391 CrossRef; (d) J. L. Vallentin., M. A. Lopez-Manchado, A. Rodriguez, P. Posadas and L. Ibarra, Appl. Clay Sci., 2007, 36, 245 CrossRef; (e) H. H. W. Moenke, in Infrared Spectra of Minerals, ed. V. C. Farmer, Mineralogical Society, London, UK, 1974, p. 365 Search PubMed.
- (a) M. A. Vicente-Rodríguez, M. Suarez, M. A. Bañares-Muñoz and J. D. D. Lopez-Gonzalez, Spectrochim. Acta, Part A, 1996, 52, 1685 CrossRef; (b) P. Komadel, in Natural Microporous Materials in Environmental Technology, ed. P. Misaelides, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1999, p. 3 Search PubMed.
- A. Choudhury, A. K. Bhowmick and C. Ong, Polymer, 2009, 50, 201 CrossRef CAS.
- Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2009, 19, 7263 RSC.
- V. M. Rusu, C. H. Ng, M. Wilke, B. Tiersch, P. Fratzl and M. G. Peter, Biomaterials, 2005, 26, 5414 CrossRef CAS.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130 CrossRef CAS.
- (a) W. J. Wu and G. H. Nancollas, Adv. Colloid Interface Sci., 1999, 79, 229 CrossRef CAS; (b) W. J. Wu and G. H. Nancollas, J. Colloid Interface Sci., 1996, 182, 365 CrossRef CAS.
- (a) X. Y. Liu and S. W. Lim, J. Am. Chem. Soc., 2003, 125, 888 CrossRef CAS; (b) J. M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382 CrossRef CAS.
- (a) R. Rodríguez-Clemente, A. López-Macipe, J. Gómez-Morales, J. Torrent-Burgués and V. M. Castaño, J. Eur. Ceram. Soc., 1998, 18, 1351 CrossRef CAS; (b) H. McDowel, T. M. Gregory and W. E. Brown, J. Res. Natl. Bur. Stand., Sect. A, 1977, 81, 273 Search PubMed; (c) J. S. Clark, Can. J. Chem., 1955, 285, 1696 CrossRef; (d) R. Kumar, K. H. Prakash, P. Cheang and K. A. Khor, Langmuir, 2004, 20, 5196 CrossRef CAS.
- J. Christoffersen and M. R. Christoffersen, J. Cryst. Growth, 1992, 121, 608 CrossRef.
- K. Onuma and A. Ito, Chem. Mater., 1998, 10, 3346 CrossRef CAS.
- (a) T. Vanorio, M. Prasad and A. Nur, Geophys. J. Int., 2003, 155, 319 CrossRef CAS; (b) M. Prasad, M. Kopycinska, U. Rabe and W. Arnold, Geophys. Res. Lett., 2002, 29, 8.
- J. C. Halpin and S. W. Tsai, AFML-TR-67-423, 1969.
- Z. Li, PhD thesis, The State University of New York, Buffalo, 1993.
Footnote |
| † Electronic supplementary information (ESI) available: XRF results of sepiolite and related biocomposites (Table S1), derivative thermogravimetry curves of HAp/sepiolite biocomposites (Fig. S1), and load-displacement curves of nanoindentation for various samples sintered at 1200 °C (Fig. S2). See DOI: 10.1039/c0nr00650e |
| This journal is © The Royal Society of Chemistry 2011 |