DOI:
10.1039/C0NR00555J
(Paper)
Nanoscale, 2011,
3, 683-692
Nanoscale morphology dependent pseudocapacitance of NiO: Influence of intercalating anions during synthesis
Received
2nd August 2010
, Accepted 22nd October 2010
First published on 22nd December 2010
Abstract
Three nano-porous NiO samples with high specific surface area were prepared by a simple hydrothermal method under homogeneous precipitation conditions using CTAB as a template and urea as the hydrolysis controlling agent. This study was done to determine the effect of different anions (acetate, nitrate and chloride) present in the precursor salts on the morphology and pseudocapacitance behavior of NiO. The samples were characterized by thermogravimetry (TG), differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD), Brunauer–Emmet–Teller (BET) isotherm and field emission scanning electron microscopy (FESEM). The final NiO samples showed different hierarchical surface morphologies and their effect on the electrochemical pseudocapacitance behavior was carefully studied by cyclic voltammetry, galvanostatic charge–discharge cycles (chronopotentiometry) and impedance spectroscopic techniques. The specific capacitance of NiO sample synthesized by NO3− ion intercalation showed higher surface area, intermediate porosity and a novel pine-cone morphology with nano-wire surface attachments. This sample exhibits the highest pseudocapacitance of 279 F g−1 at a scan rate of 5 mV s−1, calculated from the cyclic voltammetry measurements. The sample synthesized by Cl− intercalation shows a nano-flower morphology with lower surface area, porosity and pseudocapacitance behaviour. The NiO sample prepared in the presence of CH3COO− ions showed a honeycomb type surface morphology with an intermediate pseudocapacitance value but higher reversibility. The galvanostatic charge–discharge and impedance spectroscopic measurements on these NiO electrodes were consistent with CV results. The Coulombic efficiency of all the three NiO samples was found to be high (∼85 to ∼99%) after 100 galvanostatic charge–discharge cycles. This study shows that the surface morphology and porosity of NiO are strongly influenced by the anions in the precursor salts, and in turn affect significantly the pseudocapacitance behavior and the power performance of NiO powders.
1. Introduction
Electrochemical capacitors have the advantage of delivering high power in a very short duration like dielectric capacitors, and can also store high quantity of energy like rechargeable batteries. These devices are important in power-source applications such as hybrid electric vehicles, memory back-up, military and space equipment, short term power sources for mobile and day-to-day electronic devices.1–3 Supercapacitors are mainly classified as electrical double layer capacitors (EDLCs) and pseudocapacitors. EDLCs build up electrical charge at the electrode/electrolyte interface described by the Gouy–Chapman–Stern–Grahame model, and pseudocapacitors utilize the surface redox reactions of an electro-active material at specified potentials.4 Hence, knowledge of the surface characteristics of electro-active materials is crucial for improving the pseudocapacitive performance. Electronically conducting polymers,5,6carbon/carbon,7–9 and metal oxides10–13 are three main categories of materials examined for electrochemical capacitor applications. Although conducting polymers like polyaniline, polypyrrole, polythiophene and their derivatives show high gravimetric and volumetric pseudo-capacitance values, their initial performance is severely affected by limited stability of these materials during redox cycling.2Carbon-based materials with high surface area are widely used only for EDLCs applications.7 The redox transitions due to variable oxidation states in transition metal oxides make Fe3O4, MnO2, MoO3, Co3O4 and RuO2 versatile materials for pseudocapacitor applications.14,15 Among all these, the crystalline and amorphous forms of RuO2·nH2O16,17 are highly acclaimed pseudocapacitor materials for which the predicted theoretical pseudocapacitance value is 1300–2220 F g−1 and the highest experimental value reported is about 1580 F g−1 in acidic electrolyte.18,19 But RuO2·nH2O is toxic and expensive for extensive technological viability and its I–V voltage window limits its application to smaller electronic devices.20
Porous NiO as a possible electrode material for supercapacitor applications was been reported the first time by Liu and Anderson,21 and later by Srinivasan and Weidner22 with a supercapcitance value of ∼50–60 F g−1. In order to increase this value further, synthesis of nanosize NiO with hierarchical porous structures, high surface area, large pore volume and novel morphologies has been attempted by various chemical methods. These properties, combined with the well-defined electrochemical redox nature of nanostructured NiO, makes it a suitable material for pseudocapacitor applications.13,23–29 A proper pore structure of NiO is an important criterion which can enhance the overall active material–electrolyte interface and the ion transfer rate. The origin of pseudocapacitance in electroactive materials like NiO is the interfacial redox reactions which strongly depend on the grain size, morphology and porosity dependent proton transfer.13,20,29–32 There is scope to control these parameters and enhance the performance of NiO by various synthesis strategies. In particular, the role of anions on the morphological evaluation and porosity during the synthesis of NiO has not been explored. In this context, the size and overall charge of the anions which intercalate during the synthesis of NiO are expected to affect the above parameters.
Homogeneous precipitation is a simple method to control due to slow nucleation during the synthesis process. This method has been employed for the synthesis of NiO using urea as hydrolysis controlling agent.13,33,34 The hydrothermal method is an established synthesis method which produces oxide materials with good crystallinity and selective morphologies.35–37 However, a combined synthetic approach of homogeneous precipitation and hydrothermal method in presence of a shape directing agent to obtain nanostructured oxide materials is rarely employed. In this work, we have carried out the combined synthetic approach to prepare porous NiO using urea to control the hydrolysis and cetyltrimethylammonium bromide (CTAB) surfactant as a shape directing agent. As the slow nucleation process allows us to study the effect of intercalation of different anions, we have systematically investigated the effect of NO3−, Cl−, and CH3COO− anions on the physicochemical properties and pseudocapacitive behavior of NiO and tried to establish a correlation between the physicochemical properties, surface morphology and electrochemical behavior of NiO samples.
2. Experimental procedures
2.1 Materials synthesis
Analytical grade Ni(NO3)2·6H2O (99%, SD Fine, India), Ni(CH3CO2)2·4H2O (99%, SD Fine, India), NiCl2·6H2O (99.9%, SD Fine, India), cetyltrimethylammonium bromide (CTAB) (Sigma) and urea (Thomas-Baker) were used as purchased without further purification. Laboratory prepared triple distilled water was used during all the synthesis procedures. For each experiment, 20 mmol of the respective Ni salt was dissolved in 100 mL of triple distilled water and was added dropwise to 10 mmol of cetyltrimethylammonium bromide dissolved in 100 mL triple distilled water. The solution was stirred for 1 h to form a homogeneous green color solution. 40 mmols of solid urea was then added to the solution and again stirred for 3 h to get complete homogeneity. The resulting solution was transparent green in colour. The solution was then transferred to a Teflon lined stainless steel autoclave of 250 mL capacity and was subjected to heating at 120 °C in an oven, for 24 h. The autoclave was then allowed to cool to room temperature and resulting fluffy grass-green solid precipitate was separated by centrifugation at 3000 rpm. The product was then repeatedly washed with triple distilled water followed by mixture of absolute ethanol and water and finally with absolute ethanol for 5 times. The precipitate was then dried in a vacuum oven at 60 °C for 24 h. The dried sample was then heated slowly at the rate of 10 °C min−1 from room temperature to 300 °C and calcined for 3 h in flowing air to get the final product. The samples prepared using nitrate, acetate and chloride precursors before calcinations were symbolized as NiON-uc, NiOA-uc and NiOC-uc respectively and the samples after calcination were symbolized as NiO-N, NiO-A and NiO-C, respectively.
2.2 Preparation of the electrode
For evaluating the electrochemical properties of NiO samples, the working electrodes were fabricated by mixing with 75 wt% of NiO and 15 wt% of acetylene black in an agate mortar. To this mixture, 10 wt% polyvinylidene difluoride (PVdF) binder dissolved in 1-methyl-2-pyrrolidinone (NMP) was added, making a slurry. The slurry was then coated on one side of the pretreated battery-grade Ni foil (0.2 mm thick, 1 cm2 area) and dried in a vacuum oven at 60 °C for 8 h. The approximate mass of NiO coated on each electrode was ∼2.5 mg. Cyclic voltammetry (CV), chronopotentiometry (CP) and electrochemical impedance spectroscopy (EIS) measurements were carried out using a three-electrode configuration on a CHI 7081C electrochemical workstation. The Ni plate coated with NiO served as working electrode while the Pt foil (1 × 2 cm2) and Hg/HgO (1.0 M KOH) electrodes served as counter and reference electrodes, respectively. All the experiments were carried out using freshly prepared 2.0 M aqueous KOH electrolyte.
2.3 Characterization techniques
Multipoint N2 adsorption–desorption experiments were carried out on an automatic Micromeritics ASAP 2020 analyzer using Brunauer–Emmett–Teller (BET) gas adsorption method at 77 K. The samples were out-gassed at 100 °C for 2 h followed by 150 °C for 10 h in a dynamic vacuum before physisorption measurements. The specific surface areas (SBET) were calculated by BET and the pore size distribution plots were generated from the desorption branch of the isotherm by the BJH method considering the cylindrical pore model. The pore volumes were obtained from the pore size distribution data. Thermogravimetric analysis of the samples were carried out on a TA-make TGA Q500 V20.10 Build 36 instrument in air flow (20 ml min−1) with a linear heating rate of 20 °C min−1. The thermally induced phase change of the precursor samples were analyzed on a TA-make DSC Q200 instrument using open pan mode in an air atmosphere. The powder X-ray diffraction (PXRD) patterns were recorded using Bruker AXS D8 Advance diffractometer at room temperature employing Cu-Kα (λ = 0.15408 nm) radiation generated at 40 kV and 30 mA with a scan rate of 0.01° s−1 from 20 to 90°. The crystallite size of the NiO powders were approximated applying the Scherrer equation, D = Kλ/(β cosθ)38 where D is the linear dimension of the particle (particle size), K is the spherical shape factor taken as (0.89) and β is the full width at half maximum height (FWHM) of the respective peak measured graphically. The average crystallite sizes were obtained from the (111), (200) and (220) peaks. Surface morphologies of NiO samples were obtained on an FEI Quanta 200 field emission microscope. The powder samples were dispersed in ethanol and deposited on a conducting carbon tape before mounting on the microscope sample holder for analysis.
3. Results and discussions
Fig. 1(a) shows the TG curves recorded from RT to 800 °C. The uncalcined products contain both intercalated and adsorbed water molecules and the quantity may vary depending on the experimental conditions.39 Accordingly, different amounts of structural water molecules are expected to be present in the samples. The structural water is removed in a multistep process at different temperatures. From the TG curve of the NiO-N-uc sample with NO3− intercalation, it is observed that the structural water is evaporated in a single broad step, up to 205 °C and the total weight loss is ∼16%. This is followed by the decomposition step of Ni(OH)2 → NiO + H2O due to the removal of crystalline water from the sample34 and the maximum decomposition temperature is ∼290 °C. The complete loss of crystalline water amounts to ∼19%, up to 400 °C. In the other two samples with CH3COO− (NiO-A-uc) and Cl−(NiO-C-uc) intercalation, the TGA patterns are similar while the weight losses and decomposition temperatures are different. The quantitative TGA data of all the samples are presented in Table S1 of the ESI.† The total decomposition process of the precursor samples is due to dehydroxylation combined with removal of intercalated anions and surfactant molecules. The intercalated anions control the amount of water incorporated into the material during the synthesis process. This can be correlated with the difference in the interlayer spacing of different samples which is directly related to the ionic radii of the intercalated anions. When ion size increases, the amount of structural water associated with it decreases. If larger anions are intercalated, the space available for the guest water molecules decreases in the sample. Therefore the water content decreases with increase in ionic size as (Cl− < NO3− < CH3COO−).
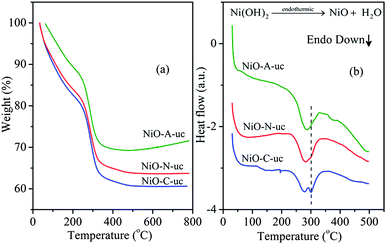 |
| | Fig. 1 (a) TG profiles and (b) the respective DSC profiles of NiO-A-uc, NiO-C-uc and NiO-N-uc samples. | |
Differential scanning calorimetry (DSC) measurements have been carried out to identify the nature of thermally induced phase changes of the uncalcined samples and the results are shown in Fig. 1(b). The DSC patterns of the three samples show different endothermic behavior, with a remnant endothermic peak at ∼300 °C. The phase change and decomposition temperatures of the three samples from TGA and DSC analysis agree well. For the NiO-A-uc sample, only a single endothermic peak is observed at ∼280 °C while NiO-C-uc and NiO-N-uc samples show doublet endothermic peaks at ∼ 280 and ∼300 °C. However, the intensity of the peak at ∼300 °C is less for NiO-N-uc sample than NiO-C-uc sample. It is known that nickel hydroxide exists in the form of hydroxyl deficient basic entities (salts) containing intercalated anions. These salts have a general composition of [Ni(OH)2−xAn−x/nyH2O where x = 0.2–0.4, y = 0.6–1 and A = chloride, nitrate, sulfate and carbonate.40 During their decomposition process, they form different crystallographic intermediate phases with differently bound inter-lamellar and crystallographic H2O molecules which are reflected as multiple endothermic peaks in the DSC analysis. The material with a smaller intercalated anion possess reduced interlayer spacing and the rate of removal of crystalline H2O molecules from it is slower, whereas with an increase in the size of the intercalated anions, the interlayer spacing increases and accordingly the rate of removal of crystalline H2O molecules increases. Therefore, the multiple phase changes are prominent in the NiO-C-uc sample with smaller interlayer spacing due to Cl− intercalation. However, for the NiO-A-uc sample with larger interlayer spacing due to the intercalation of CH3COO− ion, the rate of removal of crystalline H2O molecules is faster and no intermediate multiple phase change could be detected which is reflected in a single broad endothermic peak in the DSC pattern. For the NiO-N-uc sample with NO3− intercalation, the interlayer spacing is in the intermediate range compared to NiO-A-uc and NiO-C-uc samples. Therefore, the rate of removal of crystalline H2O molecules is slower than NiO-A-uc and faster than NiO-C-uc sample, which results in an endothermic doublet in the DSC analysis.
The wide angle XRD profiles of NiO-N-uc, NiO-C, NiO-N and NiO-A samples are shown in Fig. 2. The XRD pattern of NiO-N-uc sample shows prominent peaks at d = 7.15, 3.68, 2.68 and 1.55 Å, which are assigned to (003), (006), (101) and (110) planes of α-Ni(OH)2,41 a type of layered double hydroxide (LDH), respectively.42 The observation of a broad peak at d = 5.2 Å is the signature of β-Ni(OH)2 phase and is a result of hydrothermal ripening of α-Ni(OH)2.33 The peaks in the uncalcined precursor are broad and asymmetric in nature, which is a signature of lower crystallinity with the existence of loose and defective stacking of the crystallographic planes (turbostratic phases) of α and β-Ni(OH)2. After heating at 300 °C in air, the precursor samples entirely transform to FCC type NiO with a space group of Fm3m(225) and major peaks corresponding to (111), (200), (220), (311) and (222) reflections. The broad diffraction indicates the nanocrystallinity of the samples and the calculated crystallite sizes are ∼3–4 nm (Table S2 of the ESI†). The calculated average crystallite size of the samples decreases from NiO-C to NiO-A with increase in the size of the intercalating anion. The large size CH3COO− ion distorts the layers in the oxide product more, resulting in higher broadening of the XRD peaks.
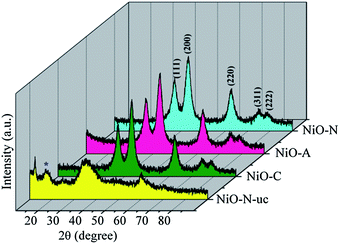 |
| | Fig. 2
XRD patterns of NiO-N-uc, and NiO-N, NiO-A and NiO-C samples calcined at 300 °C for 3 h. * is a signature of the α-Ni(OH)2 phase. | |
The N2 adsorption–desorption isotherms and pore size distribution graphs of the samples are shown in Fig. 3 and Fig. 4, respectively. All the NiO samples display irreversible type IV isotherms confirming their porous nature.43 However, the hysteresis loops are not identical implying that the porosity is different in all the samples. NiO-N and NiO-C samples exhibit H4 type hysteresis loop which is a signature of the presence of narrow slit like pores where as NiO-A sample shows an H3 type hysteresis loop owing to the existence of slit-shaped pores from the aggregations of plate-like particles. The small slopes in the p/po region of 0 to ∼0.04 (inset 1 in Fig. 3) in the adsorption branches of the isotherms indicates the presence of very small fraction of micropores in the samples.43 These micropores originate from the internal space of the porous sample (inset 2 in Fig. 3). From the lower p/po region and Horvath–Kawazoe (HK) method of micropore size distribution (Fig. S1 of the ESI†),44 it is substantiated that the fraction of micropores in the NiO-A sample is higher than that in NiO-C and NiO-N samples. The narrow micropore size distributions for all the samples are observed at a ∼1.3 nm. However the intensity of the peaks which are directly related to the volume of N2 adsorbed is found to be in the order of NiO-C < NiO-N < NiO-A which is in agreement with the increasing size of the anions of the precursor. It appears that the larger intercalating anions induce higher microporosity in the NiO structure. For all the isotherm plots, soft slopes and hysteresis loops are observed in the relative pressure (p/po) region of ∼0.1 to 0.8, which indicates the presence of large fractions of mesopores of diameter ∼2–5 nm in the samples. These mesopores are due to surface or peripheral porosity of the particles.45,46 The higher slope of the isotherm for NiO-A sample in this region demonstrates that larger CH3COO− ion induces higher mesoporosity than smaller NO3− and Cl− anions (Fig. 3). The BJH pore size distribution in Fig. 4 reveals an hierarchical multimodal porosity consisting of meso- as well as macropores in the samples. The pore size distribution in NiO-C and NiO-N samples is significantly different and narrower than the NiO-A sample. The major mesopore size distribution of all the samples is found to be around 3.5 nm. The sharp increase in the uptake of N2 at higher relative pressure (p/po>0.9) demonstrates the existence of macropores in the samples which is due to the inter-particle space (inset 2 in Fig. 3).45 Higher N2 uptake shows the presence of a large fraction of macropores in NiO-A compared to NiO-C and NiO-N samples. For the NiO-A sample, the largest macropore size is ∼34.1 nm while NiO-N and NiO-C samples show pores of ∼31 and 27.9 nm in size, respectively. The uptake of N2 in the macropore region is significantly higher for NiO-A sample suggesting the presence of higher fraction of macropores. From the macropore size distribution (above 5 nm) in Fig. 4, it can be observed that the maximum around 31 nm of NiO-N sample is relatively less intense. Therefore, the contribution of macropore volume to the total pore volume of NiO-N sample is comparatively less and the total porosity in this sample is mostly due to mesopores. But in NiO-A and NiO-C samples, the corresponding intensities of the meso- and macropores are comparable and hence the total pore volume consists of equal contribution from meso- and macropores. The quantitative structural parameters are summarized in Table 1. The surface areas of all the samples are found to be significantly higher than recently reported by Yu et al.28NiO-N sample shows the highest BET specific surface area where as NiO-A sample shows more BJH pore volume due to the variation in pore parameters. Also, the high average pore diameters for NiO-A and NiO-C samples are due to the presence of multimodal pore structures.
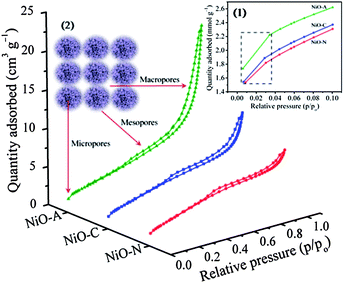 |
| | Fig. 3 BET isotherms of NiO-N, NiO-A and NiO-C samples. Inset 1 shows adsorption branches of the isotherms at p/po of 0 to 0.1. Inset 2 shows the schematic origin of the porosity in the NiO samples. | |
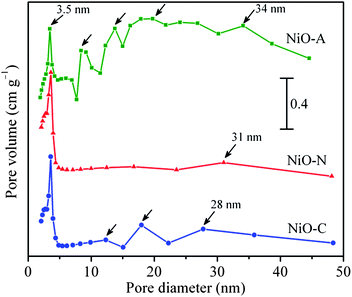 |
| | Fig. 4
Pore size distribution of NiO-N, NiO-A and NiO-C samples obtained from desorption branch of the isotherms using BJH method. | |
Table 1 Quantitative structural parameters for different NiO samples
| Sample |
Surface area, SBET/m2 g−1 |
Average pore diameter, dBJH/nm |
Total pore volume, VTOTAL/cm3 g−1 |
|
NiO-C
|
244 |
5.7 |
0.362 |
|
NiO-N
|
265 |
4.2 |
0.269 |
|
NiO-A
|
252 |
9.5 |
0.693 |
The formation of layered Ni(OH)2 involves a simple hydrolysis–precipitation process.41,47 In this process, urea acts as the hydrolysis controlling agent which decomposes to form OH− ion in aqueous conditions and further co-ordinate with Ni2+ to form Ni(OH)2. The controlled generation of OH− ions in the reaction system controls the rate of hydrolysis and crystal growth processes. Further, hydrothermal condition adds the advantage of good control over the structure and crystallization. The following reaction steps are involved in the formation of Ni(OH)214
| | 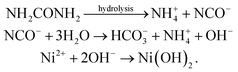 | (1) |
The solutions after the reactions were slightly basic in nature and the pH values (∼ 8.0–8.2) were much less than the pH maintained in the precipitation route to prepare β-Ni(OH)2 (i.e. pH ∼12) using aqueous ammonia. The relatively unstable α-Ni(OH)2 phase is stabilized in the lower pH medium which gets transformed to stable β-Ni(OH)2 under higher alkaline conditions.47Ni(OH)2 is the primary skeleton which after decomposition at 300 °C forms NiO with the removal of water molecules and intercalated anions. The surface morphology of all the three samples of Ni(OH)2 (NiO-A-uc, NiO-N-uc and NiO-C-uc) analyzed using a field emission gun equipped scanning electron microscope (FESEM) are shown in Fig. S2 of the ESI.† The FESEM images of the NiO samples are shown in Fig. 5. It is pronounced that the surface morphology of the respective precursor Ni(OH)2 samples have been translated very well to the final NiO products with significant structural retention. However, the porosity of the NiO samples may not quite be similar to the respective Ni(OH)2 precursors, but as the porosity of NiO samples are from the decomposition of water and intercalating anions in Ni(OH)2, it is reasonable to say that the intercalating anions have adequate indirect control over the porosity of NiO. The low magnification FESEM micrographs of NiO samples in Fig. 5 show almost uniform sized and porous ball like structures, but at higher magnification, the micrographs show differently porous surface morphologies. This is confirmed from the HRTE Micrographs of the respective samples shown in Fig. 6. It is known that crystallographic shapes are composed of crystallographic planes, which are further related to the surface energies. It is also known that organic and inorganic additives adsorb onto certain crystallographic planes and modify the relative order of surface energies during the crystal growth process.48 This preferential adsorption lowers the surface energy and directs the formation of a particular morphology of the material. Different anionic species can also adsorb on different crystallographic planes to lower the respective surface energies and give different morphology to the material. The NiO-A sample shows a honeycomb like macroporous structure made up of hierarchically oriented hairy plate like structures as shown in Fig. 5(a) and 6(a). The larger CH3COO− ion intercalation induces the highly porous honeycomb morphology to the material. The high magnification microscopic images of NiO-N samples in Fig. 5(b) and 6(b) show a unique pine-cone like surface morphology consisting of oriented and densely packed nano-fiber structures. This indicates that the NiO-N particles are, in fact, self-assembled NiO nano-fibres. Microscopic images of NiO-C sample in Fig. 5(c) and 6(c) show flower like morphology composed of oriented and self assembled micropetals. The mechanism for the formation of these superstructures is not clear,49 but it is likely that the final structures are primary aggregations of nanosized particles nucleated in a supersaturated solution although the kinetic pathway of crystallization involves a multistep process50 and factors like van der Waals forces, hydrophobic interactions, crystal-face attraction, electrostatic and dipolar fields, and hydrogen bonds immensely contribute to the final morphology of the material.51,52 It should be noted that the mesoporosity in the NiO samples may also be due to the intrinsic crystal contraction, a concept proposed by Lou et al.53 in the case of Co3O4.
 |
| | Fig. 5
FESEM images of NiO-N, NiO-A and NiO-C samples showing the pine-cone, honeycomb and flower-like morphologies respectively. | |
 |
| | Fig. 6
HRTEM images of (a) NiO-A (b) NiO-N and (c) NiO-C samples. | |
The presence of CTAB is crucial for hierarchical structure and porosity of NiO. Parallel experiments of synthesizing NiO under same experimental conditions without the use of surfactant produced a very low surface area with no specific morphology, as recently reported by Zheng et al.54
3.1 Electrochemical properties
3.1.1
Cyclic voltammetry
.
The cyclic voltammograms (CVs) of NiO-C, NiO-N and NiO-A samples performed at a sweep rate of 5 mV s−1 and in a potential range of −0.2 to 0.6 V (vs.Hg/HgO) are illustrated in Fig. 7 and the calculated parameters are summarized in Table 2. The NiO electrode has been stabilised initially for 25 CV cycles before the final measurements. The CVs of all samples show almost similar patterns and their shape reveals a pseudo-capacitance behaviour, characteristic of a redox mechanism.26 The anodic peak at the positive current density and cathodic peak at the negative current density are the result of oxidation of NiO to NiOOH (charging) and reduction of NiOOH to NiO (discharging) processes, respectively. The potentials at which the respective oxidation and reduction take place are termed as oxidation potential (EO) and reduction potential (ER), respectively. During the cathodic scanning, peak at the higher potential is due to the oxygen-evolution reaction and the corresponding potential is termed as oxygen-evolution potential, EOE. The following redox processes take place exclusively during the CV analysis.| | 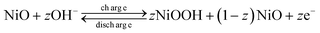 | (2) |
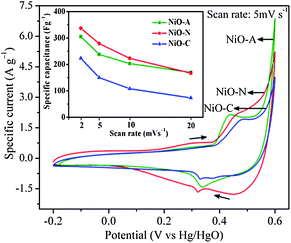 |
| | Fig. 7
Cyclic voltammograms of NiO-N, NiO-A and NiO-C electrodes measured at a scan rate of 5 mV s−1. | |
Table 2
Reduction potential (ER), oxidation potential (EO), oxygen evolution potential (EOE), (EO − ER) and (EOE − EO) for different NiO
| Sample |
E
R/mV |
E
O/mV |
E
OE/mV |
E
O − ER/mV |
E
OE − EO/mV |
|
NiO-C
|
330 |
489 |
602 |
159 |
113 |
|
NiO-N
|
322 |
463 |
602 |
141 |
139 |
|
NiO-A
|
337 |
442 |
602 |
103 |
160 |
The cathodic sweeps of CV curves are not completely symmetric to their corresponding anodic sweeps, which indicates some irreversibility in the redox process.4 Practically pseudo-capacitors involving Faradaic redox reactions suffer polarization and the ideal reversibility cannot be realized kinetically for the positive and negative sweeps.4,12 Ohmic resistance due to electrolyte diffusion within the porous electrode also contributes to the kinetic irreversibility of the redox reaction. The ΔE (EO − ER) value is also a measure of reversibility in the redox reaction; a smaller value corresponds to better reversibility and vice versa. Among the three samples, NiO-A shows the highest reversibility (ΔE = 103 mV) while NiO-C shows the least reversibility (ΔE = 159 mV).
This trend is in agreement with the size of the macropores in the samples. NiO-A sample having a honeycomb like structure and large pore volume allows easy diffusion of OH− ions for higher reversibility during the redox reactions. However the NiO-N sample with a fibrous surface is poorly feasible for diffusion of OH− ions. The flower type surface morphology of NiO-C sample does not promote easy diffusion of the ions as it has the least reaction reversibility. The intensities of EO and ER peaks are important parameters to check the energy density of the electrode material. Higher the intensity of the peaks show better energy density of the material. It is observed from Fig. 7 that the NiO-A sample possesses the highest and NiO-C sample possesses the lowest energy density. This may be due to the ideally porous surface morphology of the NiO-A sample. The difference between the oxygen evolution potential and the oxidation potential, EOE − EO, is a measure of the performance of the NiO electrode. The NiO-A electrode has higher EOE − EO value (∼160 mV) and hence better electrochemical cycling properties than NiO-N (139 mV) and NiO-C (113 mV). The higher EOE − EO value of NiO-A also reveals that Ni2+ of the honeycomb electrode completely oxidizes to Ni3+ before oxygen evolution takes place.55 The specific capacitance (Cs) values of the samples estimated from CV curve is shown graphically in Fig. S3 of the ESI†. The Cs is calculated graphically by integrating the area under the I–V curve and then dividing it by the sweep rate v (V s−1), the mass of NiO (w) and the potential window (Va to Vc), using the equation:
| | 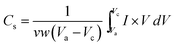 | (3) |
where
I (A) is cathodic or anodic current and Δ
V (
V) is the applied potential window. The NiO-N sample shows the highest current response and capacitance values while NiO-C exhibits the lowest values. The calculated specific capacitance values for NiO-A, NiO-N and NiO-C samples are 196, 279 and 156 F g
−1, respectively, at a scan rate of 5 mV s
−1. So, the NiO-N sample provides a larger electroactive surface area for redox reactions and charge storage compared to NiO-A and NiO-C samples. This is due to better micro-structural properties such as a higher specific surface area, higher micro and mesopore volumes and hierarchical fibrous surface morphology. This sample also has suitable average pore diameter of 4.2 nm for superior electrochemical performance and facilitates better OH
− propagation and high current loads.
20,57 More OH
− ions can preferentially access the surface of the fibrous structure for maximum Faradaic reaction than the honeycomb and nano-flower structures, shown in the pictorial representation (
Scheme 1).
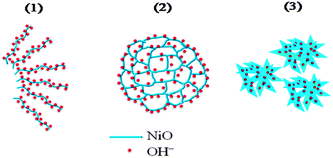 |
| | Scheme 1 Pictorial representation of (1) fibrous NiO-N, (2) honeycomb NiO-A and (3) flower-like NiO-C structures showing accessibility to OH− ions. | |
It is therefore expected that in the surface redox process, a higher fraction of nickel sites (z in eqn (2)) are involved in the NiO-N sample with fibrous structure than NiO samples with honeycomb and flowery structures. The value of “z” in eqn (2) can be estimated from the specific capacitance value of the NiO by using the expression56
| | 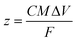 | (4) |
with a potential window (Δ
V) of 0.8 V, molecular weight (
M) of 74.692 g, the Faraday constant (96
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
487 C/equiv.) and the calculated specific capacitance values (
C) for NiO samples from
Fig. 7, the “
z” values obtained for NiO-N, NiO-A and NiO-C samples are 0.1728, 0.1214 and 0.0966, respectively. An estimated 17.3% of NiO-N, 12.1% of NiO-A and 9.7% of NiO-C sites participate in the redox reactions which are significantly higher than the values reported on NiO films.
56 Accordingly the hierarchical surface structures expose more Ni
2+ sites for sufficient redox reactions. The inset of
Fig. 7 shows the specific capacitance values of the samples as a function of scan rates of 2, 5, 10 and 20 mV s
−1. In the NiO-A sample, there is a marginal decrease in specific capacitance at higher scan rates compared to NiO-N and NiO-C samples. This indicates a higher power performance of honeycomb structured NiO-A sample than of the NiO-N and NiO-C samples.
58 This is attributed to the rigid honeycomb structure of the NiO-A sample where the morphology is unaffected by high scan rates, while the flaking-off of the loose flakes takes place at higher scan rates for NiO-N and NiO-C samples. So, the pine cone structured NiO-N sample gives higher specific capacitance, whereas the rigid honeycomb structure shows higher power performance. These results suggest morphology dependent redox reactions and power performance during the charge-storage process.
3.1.2
Chronopotentiometry
.
To evaluate the cyclic and service life of the NiO samples, chronopotentiometry (CP) measurements were carried out at a fixed mass normalized charge and discharge current densities of 200 mA g−1 and the results are shown in Fig. 8. The non-linear discharge curves of each CP graphs shows a small linear variation of the potential from 0.05 to ∼0.3 V with respect to time, which indicates the double-layer capacitance behaviour due to the charge separation between the electrode and electrolyte interface. The variation of the potential from ∼0.3 to 0.55 V is due to typical pseudo-capacitance behaviour resulting from the electrochemical adsorption/desorption or redox reaction at the electrode–electrolyte interface.59 The overall shapes of the chronopotentiometric curves suggest the pseudocapacitance behaviour of NiO electrodes, which is in agreement with CV analysis. The charging and discharging curves of the samples are not completely symmetrical due to the certain kinetic irreversibility of the OH− ions on the porous NiO surface during the redox reaction.4,12 This is also supported by the dissimilarity in the charging (tC) and discharging time (tD). However, the NiO-A sample shows higher symmetry in the charging and discharging curves and lower (tC − tD) values which can be attributed to better reversibility in the honeycomb structure. The honeycomb surface morphology is suitable to act as perfect “OH− ion-buffering reservoir” resulting in a shorter diffusion path length of the ions for higher diffusion rates, and significantly faster electronic kinetics for maximum reversible redox reactions.60 The time durations of charge and discharge cycles for different samples are not the same at the same current density, which suggests a difference in their specific capacitance values. The specific capacitance (Cs) values can be calculated from the galvanostatic charge–discharge cycles using the following equation61| | 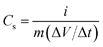 | (5) |
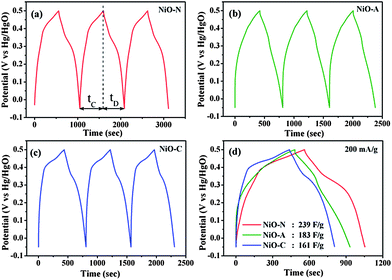 |
| | Fig. 8 Galvanostatic charge–discharge cycles (chronopotentiometry) of (a) NiO-N, (b) NiO-A and (c) NiO-C electrodes measured at a current density of 200 mA g−1. The comparison of their first charge–discharge cycles is also shown in (d). | |
In this equation, i is the current (A) applied for time t (s), m is the mass of nickel oxide (g) and ΔV/Δt is the slope of the discharge curve (V s−1). The comparison of charge–discharge cycles of NiO-N, NiO-A and NiO-C samples measured at 200 mA g−1 are presented in Fig. 8(d) and the calculated specific capacitance values are found to be 239, 183 and 161 F g−1, respectively. These values show similar trends as those obtained from cyclic voltammetry (Fig. 7). The variation in specific capacitance values of different samples is due to the difference in their microstructural properties. The NiO-N sample with a fibre-like structure facilitates better electrolyte contact and ion diffusion rate and larger BET surface area provides more accessible electroactive surface sites for charge storage. However, the NiO-A sample, due to its honeycomb morphology, facilitates the movement of OH− ions through its channels and exhibits better reversibility than the other samples. The NiO-C sample with a flower like-morphology has a lower surface area and does not possess large electroactive surface sites and suitable porosity for good electrolyte contact, resulting in a lesser charge storage capacity and reversibility. For practical use of an electrochemical capacitor, life-cycle stability is very important. Fig. 9 illustrates the relationship between capacity retention and galvanostatic charge–discharge cycling at 200 mA g−1 for all the three samples. Although a minimum of 1000 galvanostatic charge–discharge cycles are required for practical applications, we have studied 100 repetitive charge–discharge cyclic processes for the preliminary information on the stability and performance of different microstructured NiO samples.62 All the samples show reasonably good cyclic stability. The pseudocapacitance values of NiO-N, NiO-A and NiO-C samples after 100 charge–discharge cycles are found to be 221, 175 and 142 F g−1, respectively. The capacitance retention of the NiO-A sample is the highest at 95.6% followed by NiO-N and NiO-C samples at 92.4 and 88.3%, respectively.
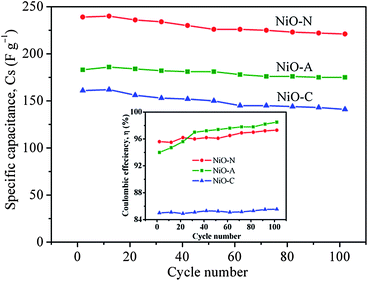 |
| | Fig. 9 The specific capacitance values of NiO-N, NiO-A and NiO-C electrodes as a function of cycle number. Inset shows the respective Coulombic efficiency. | |
This is due to the fact that the rigid honeycomb structure undergoes least structural modification or degradation or crystallographic changes
15 during the repeated insertion/
extraction of OH
− ions into/from the NiO-A sample. However, the flaky NiO-N and NiO-C samples tend to undergo easy physical modification by flaking off at higher applied current densities for longer times and thus show lower cyclic stability. Small increases in specific capacitance values were observed for all the samples up to the first 10–12 charge–discharge cycles due to some initial surface
activation of their porous structures. The efficacy of charge transfer in a system to facilitate an electrochemical reaction is termed the Coulombic efficiency (
η) and the higher value of “
η” suggests the requirement of lower energy to complete the reaction and make the process more feasible. The Coulombic efficiency can be calculated from the following relation:
| | 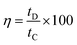 | (6) |
where
tD and
tC represents galvanostatic discharging and charging time, respectively. The Coulombic efficiency is found to be the highest for NiO-A sample and lowest for NiO-C samples after 100 charge–discharge cycles (Inset of
Fig. 9). The calculated Coulombic efficiencies of NiO-A, NiO-N and NiO-C samples measured from the 102
nd charge–discharge cycle are ∼98.5, ∼97.3 and ∼85.6%, respectively. The observed higher Coulombic efficiency of NiO-A sample is due to the higher reversibility of the material which is consistent with the CV and CP studies. It is interesting to observe a rapid increase in the Coulombic efficiency value of NiO-A sample up to 20–25 charge–discharge cycles followed by a slight increase up to 100 cycles. The increase in Coulombic efficiency value represents an increase in kinetic reversibility with cycling. This should be attributed to pore opening and
activation of the hierarchically porous NiO-A sample during the Faradaic reaction in a galvanostatic charge–discharge processes. However, the NiO-N and NiO-C samples do not exhibit increasing trends in Coulombic efficiency with cycling and show relatively poor
activation and kinetic reversibility.
These observations demonstrate that the honeycomb morphology is electrochemically preferred over the fibrous surface structure.
3.1.3 Electrochemical impedance spectral (EIS) measurements.
For electrochemical supercapacitors to be used as power devices, the active electrode material should have lower electronic resistance (or higher electrical conductivity) to achieve higher capacitance values. The AC impedance technique is employed in this study to quantitatively evaluate the relative electronic resistance and their capacitance values in various microstructured NiO samples. The EIS measurements are performed between 0.1 to 105 Hz. The Nyquist plots (imaginary part, Z′′ versus real part, Z′) of the NiO-A, NiO-N and NiO-C samples are presented in Fig. 10. A simple circuit model to evaluate the EIS spectra is shown in the inset of Fig. 10.12,63 The impedance behaviors of the samples are analyzed in the high, medium and low-frequency regions which demonstrate different processes occurring in different frequency regions. The two partial semicircles at high and medium frequency regions are often attributed to the different charge propagation phenomena in the system as a result of Faradaic reactions. The expanded view of the semicircles at higher frequency region is shown in Fig. 11. These are characteristic of the processes occurring at the oxide–electrolyte interface which can be modelled as a double-layer capacitor Cd in parallel with an charge–transfer resistor, Rict. The Rict is the resistance due to some discontinuity in the charge transfer process at the solid oxide/liquid electrolyte interface which arises from the difference in conductivity between the solid oxide (electronic conductivity) and the aqueous electrolyte phase (ionic conductivity). The Rict for NiO-N sample has the least value followed by NiO-A and NiO-C samples. This observation illustrates the higher conductivity of NiO-N sample which is due to its thin fiber type morphology as well as higher electroactive surface area. The second partial kinetic semicircles at medium frequency regions correspond to the charge-transfer resistance due to Faradaic redox processes in the system which involves the exchange of OH− ions. This is associated with the surface phenomena of the porous NiO electrode. This impedance characteristic can be modeled as a film impedance due to the Faradaic redox processes involving electron hopping in the NiO particle and OH− ion diffusion. This is assumed to be consisting of a film capacitor, Cf, in parallel with an electron-transfer resistor, Rect. The characteristics resistance in this medium frequency region is lowest for the NiO-N sample whereas the NiO-A and NiO-C samples show higher resistance. It is known that the charge-transfer resistance is a surface property of the porous electrode which is related to the electroactive surface area. It is a combination of electrolyte accessible area and electrical conductivity of the electrode material. The larger the electroactive surface area, the lower the charge-transfer resistance.64 The NiO-N sample, having higher surface area and long thin flake like morphology, has higher conductivity and permits higher electrolyte accessibility for enhanced Faradaic reactions. The charge transfer resistance in the medium frequency region follows the order: NiON < NiOA < NiO-C which is in agreement with the order of BET surface areas of the samples. So, as the surface area is higher, more OH− ions have the access to the surface of the material which results in a lower charge transfer resistance. At a lower frequency region of the EIS plot in Fig. 10, the plot is almost linear, which corresponds to the Warburg impedance, W, and is described as diffusive resistance of the OH− ion within the NiO electrode pores. Generally a straight vertical line along the imaginary axis Z′′ is observed for an ideally polarized system where as in a real capacitor, a finite slope appears in this line which represents the diffusive resistance of electrolyte in electrode pores and ion diffusion in host materials as the diffusion of OH− is not same in the liquid electrolyte and solid electrode materials. The almost-straight vertical line for the NiO-N sample demonstrates the lower diffusion resistance due to its highly porous and fibrous morphology. This leads to easy access of the OH− ions as shown in the Scheme 1. NiO-N sample also shows a lower combination of ionic resistance of electrolyte, intrinsic resistance of substrate and contact resistance at the active material/current collector interface.65 The experimental impedance data are converted to specific capacitance (Cs) using the following equation:66| | 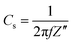 | (7) |
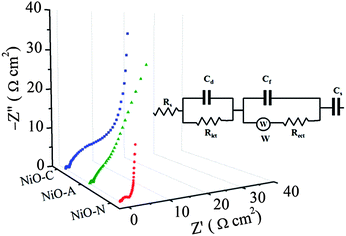 |
| | Fig. 10 Complex plane impedance plots (Nyquist plots) of NiO-N, NiO-A and NiO-C electrodes. Inset shows the equivalent circuit. | |
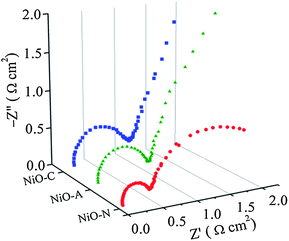 |
| | Fig. 11 Complex plane impedance plots (Nyquist plots) of NiO-N, NiO-A and NiO-C electrodes at higher frequency region. | |
The specific capacitance calculated from impedance analysis is plotted against frequency, f, and is shown in Fig. S4 of the ESI.† The estimated specific capacitance value of the NiO-N sample is found to be the highest at all frequencies, followed by the NiO-A and NiO-C samples. The specific capacitances were measured to be 355, 262 and 194 F g−1 at 0.01 Hz. This trend is in agreement with the resistances observed for the respective samples in the impedance plots. This result is also in concurrence with the CV and CP analysis.
Conclusions
In summary, a hydrothermal-homogeneous precipitation method using urea and precursors with different counter anions of Ni salts has been pursued to synthesise three distinctive morphologies of NiO superstructures with hierarchical meso- and macroporosity. Self-aggregation is believed to be the primary parameter for the formation of hierarchical morphologies. Cyclic voltammetry studies demonstrate the morphology-dependent pseudocapacitance behaviour as well as power performance of NiO samples. It is found that the specific capacitance of the NiO-N sample with a fibrous-pine cone morphology has a higher specific capacitance, but the honeycomb-structured NiO-A sample shows a higher power performance capability. The chronopotentiometric studies shows a higher pseudocapacitance for the NiO-N sample and a better reversibility for the NiO-A sample. The NiO-C sample with a flower-like surface morphology is found to have the lowest specific surface area and, accordingly, shows lower specific capacitance and reversibility. The Coulombic efficiency of the samples measured after 100 galvanostatic charge–discharge cycles are found to be 98.5, 97.3 and 85.6% for NiO-A, NiO-N and NiO-C samples, respectively. The impedance analysis shows the charge storage process is hindered by a combination of ionic resistance of the KOH electrolyte, intrinsic resistance of the NiO substrate and the contact resistance at the active material/Ni current collector interface. However, the NiO-N sample is found to have the least overall resistance followed by NiO-A and NiO-C samples, and this trend is reflected in the specific capacitance values calculated from the EIS measurements. This work throws light on the pseudocapacitance behaviour and performance of an electroactive material which are controlled not only by the surface area, pore size and pore volume, but also by the surface morphologies.
Acknowledgements
This work is financially supported by the Defence Research and Development Organisation (DRDO), Ministry of Defence, New Delhi, through grant No. ERIP/ER/0600319/M/01/1052.
Notes and references
- R. A. Huggins, Solid State Ionics, 2000, 134, 179 CrossRef CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483 CrossRef CAS.
- J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef CAS.
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539 CAS.
- T. C. Girija and M. V. Sangaranarayanan, J. Power Sources, 2006, 156, 705 CrossRef CAS.
- P. Novak, K. Muller, K. S. V. Santanam and O. Hass, Chem. Rev., 1997, 97, 207 CrossRef CAS.
- C. Yu, C. Masarapu, J. Rong, B. Wei and H. Jiang, Adv. Mater., 2009, 21, 4793 CrossRef CAS.
- A. L. M. Reddy and S. Ramaprabhu, J. Phys. Chem. C, 2007, 111, 7727 CrossRef CAS.
- S. R. C. Vivekchand, C. S. Rout, K. S. Subramanyam, A. Govindaraj and C. N. R. Rao, J. Chem. Sci., 2008, 120, 9 CrossRef CAS.
- J. W. Long, K. E. Swider, C. I. Merzbacher and D. R. Rolison, Langmuir, 1999, 15, 780 CrossRef CAS.
- J. Rajeswari, P. S. Kishore, B. Viswanathan and T. K. Varadarajan, Electrochem. Commun., 2009, 11, 572 CrossRef CAS.
- S. K. Meher, P. Justin and G. Ranga Rao, Electrochim. Acta, 2010, 55, 8388 CrossRef CAS.
- P. Justin, S. K. Meher and G. Ranga Rao, J. Phys. Chem. C, 2010, 114, 5203 CrossRef CAS.
- K. W. Chung, K. B. Kim, S.-H. Han and H. Lee, Electrochem. Solid-State Lett., 2005, 8, A259 CrossRef CAS.
- P. Ragupathy, D. H. Park, G. Campet, H. N. Vasan, S. Hwang, J. Choy and N. Munichandraiah, J. Phys. Chem. C, 2009, 113, 6303 CrossRef CAS.
- J. P. Zheng, Electrochem. Solid-State Lett., 1999, 2, 359 CrossRef CAS.
- C.-C. Hu, W.-C. Chen and K.-H. Chang, J. Electrochem. Soc., 2004, 151, A281 CrossRef CAS.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6, 2690 CrossRef CAS.
- I.-H. Kim, J.-H. Kim and K.-B. Kima, Electrochem. Solid-State Lett., 2005, 8, A369 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- C. K-Liu and M. A. Anderson, J. Electrochem. Soc., 1996, 143, 124 CrossRef CAS.
- V. Srinivasan and J. W. Weidner, J. Electrochem. Soc., 1997, 144, L210 CAS.
- H. Pang, Q. Lu, Y. Zhang, Y. Lia and F. Gao, Nanoscale, 2010, 2, 920 RSC.
- M.-S. Wu, Y.-A. Huang, J.-J. Jow, W.-D. Yang, C.-Y. Hsieh and H.-M. Tsai, Int. J. Hydrogen Energy, 2008, 33, 2921 CrossRef CAS.
- H. Pang, Q. Lu, Y. Lia and F. Gao, Chem. Commun., 2009, 7542 RSC.
- J.-W. Lang, L.-B. Kong, W.-J. Wu, Y.-C. Luo and L. Kang, Chem. Commun., 2008, 4213 RSC.
- R. J. Brodd, J. Electrochem. Soc., 2004, 151, K1 CrossRef CAS.
- C. Yu, L. Zhang, J. Shi, J. Zhao, J. Gao and D. Yan, Adv. Funct. Mater., 2008, 18, 1544 CrossRef CAS.
- W. Zhou, M. Yao, L. Guo, Y. Li, J. Li and S. Yang, J. Am. Chem. Soc., 2009, 131, 2959 CrossRef CAS.
- X. H. Huang, J. P. Tu, X. H. Xia, X. L. Wang, J. Y. Xiang, L. Zhang and Y. Zhou, J. Power Sources, 2009, 188, 588 CrossRef CAS.
- S. W. Oh, H. J. Bang, Y. C. Bae and Y.-K. Sun, J. Power Sources, 2007, 173, 502 CrossRef CAS.
- C. Yuan, S. Xiong, X. Zhang, L. Shen, F. Zhang, B. Gao and L. Su, Nano Res., 2009, 2, 722 CrossRef CAS.
- G. J. A. A. S. Illia, M. Jobbgy, A. E. Regazzoni and M. A. Blesa, Chem. Mater., 1999, 11, 3140 CrossRef.
- W. Xing, F. Li, Z. F. Yan and G. Q. Lu, J. Power Sources, 2004, 134, 324 CrossRef CAS.
- L.-X. Yang, Y.-J. Zhu, H. Tong, Z.-H. Liang and W.-W. Wang, Cryst. Growth Des., 2007, 7, 2716 CrossRef CAS.
- Y. Shao, J. Sun and L. Gao, J. Phys. Chem. C, 2009, 113, 6566 CrossRef CAS.
- C. Wu, W. Xie, M. Zhang, L. Bai, J. Yang and Y. Xie, Chem.–Eur. J., 2009, 15, 492 CrossRef CAS.
- B. Mani and J. P. de Neufville, J. Electrochem. Soc., 1988, 135, 800 CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- M. Rajamathi and P. V. Kamath, J. Power Sources, 1998, 70, 118 CrossRef CAS ; and references there in.
- P. Jeevanandam, Yu. Koltypin and A. Gedanken, Nano Lett., 2001, 1, 263 CrossRef CAS.
- L. Dong, Y. Chu and W. Sun, Chem. Eur. J., 2008, 14, 5064 CrossRef CAS.
-
F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids, Academic Press, London, 1999 Search PubMed.
- S. U. Rege and R. T. Yang, AIChE J., 2000, 46, 734 CrossRef CAS.
- M. Rose, W. Bohlmann, M. Sabo and S. Kaskel, Chem. Commun., 2008, 2462 RSC.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67 RSC.
- L. Xu, Y.-S. Ding, C.-H. Chen, L. Zhao, C. Rimkus, R. Joesten and S. L. Suib, Chem. Mater., 2008, 20, 308 CrossRef CAS.
- M. J. Siegfried and K.-S. Choi, Adv. Mater., 2004, 16, 1743 CrossRef CAS.
- J. Park, V. Privman and E. Matijevic, J. Phys. Chem. B, 2001, 105, 11630 CrossRef CAS.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576 CrossRef.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769 CrossRef CAS.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, J. Mater. Chem., 2008, 18, 4397 RSC.
- Y.-Z. Zheng, H.-Y. Ding and M.-L. Zhang, Mater. Res. Bull., 2009, 44, 403 CrossRef CAS.
- W. Li, S. Zhang and J. Chen, J. Phys. Chem. B, 2005, 109, 14025 CrossRef CAS.
- V. Srinivasan and J. W. Weidner, J. Electrochem. Soc., 2000, 147, 880 CrossRef CAS.
- A. B. Fuertes, G. Lota, T. A. Centeno and E. Frackowiak, Electrochim. Acta, 2005, 50, 2799 CrossRef CAS.
- H.-Q. Wang, Z.-S. Li, Y.-G. Huang, Q.-Y. Li and X.-Yu. Wang, J. Mater. Chem., 2010, 20, 3883 RSC.
- W. Sugimoto, H. Iwata, Y. Yasunaga, Y. Murakami and Y. Takasu, Angew. Chem., Int. Ed., 2003, 42, 4092 CrossRef CAS.
- D.-W. Wang, F. Li, M. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS.
- M.-S. Wu, Y.-A. Huang and C.-H. Yang, J. Electrochem. Soc., 2008, 155, A798 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS.
- C.-C. Hu, K.-H. Chang and T.-Y. Hsu, J. Electrochem. Soc., 2008, 155, F196 CrossRef CAS.
- M. S. Wu, Appl. Phys. Lett., 2005, 87, 153102 CrossRef.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109 CrossRef.
-
B. E. Conway, in Electrochemical Supercapacitors, Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated TGA and XRD results; pore size distribution from HK method; SEM pictures of uncalcined samples; graphically plotted capacitance values from CV and impedance measurements. See DOI: 10.1039/c0nr00555j |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.