Magnetic nanobeads decorated by thermo-responsive PNIPAM shell as medical platforms for the efficient delivery of doxorubicin to tumour cells†
Smriti R.
Deka‡
a,
Alessandra
Quarta‡
ab,
Riccardo
Di Corato
b,
Andreas
Riedinger
a,
Roberto
Cingolani
a and
Teresa
Pellegrino
*ab
aIstituto Italiano di Tecnologia, via Morego 30, 16163, Genova, Italy. E-mail: teresa.pellegrino@unisalento.it; Fax: +39 0832 298 230; Tel: +39 0832 298 214
bNational Nanotechnology Laboratory of CNR-NANO, via per Arnesano km 5, 73100, Lecce, Italy
First published on 16th November 2010
Abstract
Medical nanoplatforms based on clusters of superparamagnetic nanoparticles decorated with a PNIPAM thermo-responsive shell have been synthesized and used as drug carriers for doxorubicin (DOXO), a common chemotherapeutic agent. The nanosystem here developed has a total diameter below 200 nm and exploits the temperature responsive behaviour of the PNIPAM polymeric shell for the controlled loading and release of DOXO. The system has been tested in vitro on tumour cells and it clearly demonstrates the effectiveness of drug polymer encapsulation and time-dependent cell death induced by the doxorubicin release. Comparative cellular studies of the DOXO loaded nanoplatform in the presence or absence of an external magnet (0.3 T) showed the synergic effect of accumulation and enhanced toxicity of the system, when magnetically guided, resulting in the enhanced efficacy of the system.
Introduction
In the last decades, remarkable progress has been accomplished in the development of new polymer based functional materials, showing stimuli-responsive behavior.1–4 These “smart” polymers have found applications in different fields, including the pharmaceutical area.5,6 In this field, the design of “intelligent” drug delivery systems requires the preparation of carriers suitable for hosting, protecting, transporting the embedded drug and for controlling its release to the target site, under the action of a defined physical stimulus, such as the cellular pH, the local temperature, or the tumor reducing environment.On the other hand, the manipulation and the delivery of the nanocontainer and whatever it is associated to it, towards a selected area (a tumor or a damaged tissue) could be certainly enhanced and facilitated if superparamagnetic nanoparticles are included within the carrier systems.7 The system becomes magnetically active upon the application of an external magnet as it will accumulate to the site where the magnet is applied, while it will be re-dispersed in solution upon the removal of the magnet. Additionally, magnetic nanoparticles, exposed to appropriate radio frequencies, could generate heat that can be exploited for hyperthermia treatment to kill tumor cells in cancer therapy or provide the external stimulus, the local heat, necessary to open the nanocontainer and release the drug.8–10
It is then understandable while active research efforts are devoted to the combination of magnetic nanoparticles with stimuli-responsive polymers. Recently, we have reported on the exploitation of acidic pH-responsive nanogel as cargo for the controlled loading and release of pH-mediated iron oxide nanoparticles (IONPs) and at the same time of short oligonucleotides (which could be the therapeutic agents in some gene-therapy).11
Different groups have been working on the growth of thermo-responsive polymers on the surface of IONPs.7,12–17Poly(N-isopropylacrylamide), PNIPAM for instance is a linear polymer that experiences a conformational change from a random coil to a collapsed state at a critical temperature, also known as lower critical solution temperature (LCST), which is equal to 32 °C.18,19 In the case of the PNIPAM-shell on IONPs, it has been shown that the polymer transition from a coil to a shrinked state at the IONP surface promotes the inter-particle aggregation and consequently the aggregates reply faster to the magnet and can be easily separated. This behavior has been exploited to trigger the separation of individual magnetic nanoparticles from the solution phase, which is still difficult to be achieved.20,21
Indeed, nanocrystals of iron oxide with dimension of the order of 10 nm have a low magnetization per particle, and thus it is difficult to separate them from the solution or to control their movement in blood by using moderate magnetic fields. This limits their use in separation and drug delivery. Increasing the nanocrystal size increases the saturation magnetization, but also induces the superparamagnetic–ferromagnetic transition (at a domain size of ca. 30 nm for Fe3O4), so that nanocrystals are no longer dispersible in solution.22,23 The PNIPAM shell on top of IONPs proposed here proved to trigger in a controlled manner the nanoparticles aggregation, thus maintaining the colloidal stability of superparamagnetic nanoparticles when the temperature is kept below 32 °C.20,24
The above mentioned superparamagnetic nanoparticles coated with PNIPAM have been also exploited for the drug encapsulation,25 however, the temperature-mediated inter-particle aggregation might represent a problem for their exploitation as in vivodrug delivery carriers.
Recently, secondary structures of magnetic nanocrystals obtained by controlled clustering of magnetic nanocrystals have been proposed as a valuable alternative to individual IONPs with potential as drug carriers. The clustering of magnetic nanoparticles offers the advantage to increase the total magnetization of the system while retaining the superparamagnetic feature of the individual magnetic nanoparticles: the magnetic clusters can be easily dispersed in solution when the magnet is removed. However, their use as magnetic carriers for drug delivery requires a precise control of the size in order to obtain stable colloidal nanoclusters, (from now on referred to as nanobeads (NBs)) which are at the same time small enough to avoid RES (reticuloendothelial system) sequestration.26
In this frame, we have recently reported a procedure to cluster, in a controlled manner, surfactant coated magnetic nanoparticles by enwrapping several iron oxide nanoparticles within a polymer shell of poly(maleic anhydride-alt-1-octadecene).27 The procedure allows to precisely tune the size, the shape and the number of IONPs per bead and to modulate the magnetic response of the resulting nanobeads (NBs). In the present work, to engineer appropriate magnetically guided carriers we have first decorated the nanobead surface with an additional layer of a thermo-responsive polymer based on PNIPAM, then we have exploited this shell for the temperature-controlled loading and release of doxorubicin, a chemotherapeutic agent. The system was tested on tumour cells and these studies allow us to elucidate the main features of our magnetic temperature-responsive nanocarriers.
In this work we demonstrate that the concept of magnetic cluster-like beads embedded in a PNIPAM shell has a great potential in the drug delivery of chemotherapeutics to tumour cells.
Results and discussion
Preparation of PNIPAM–NBs and their characterization
The procedure for the synthesis of PNIPAM–NBs is illustrated in Scheme 1. By using a protocol recently reported by us,27 we first prepared clusters of iron oxide nanoparticles, the so called starting NBs, by enwrapping multiple iron oxide nanoparticles (6 nm in diameter) within an amphiphilic polymer named poly(maleic anhydride-alt-1-octadecene) (PC18). In order to grow the PNIPAM shell, the anhydride groups of the polymer at the bead surface could be further processed. For this purpose we first linked short priming ligands, the 2-aminoethylmethacrylate molecules (AEM) to the nanobead surface, the primary amino group of the AEM forms an amide bond with the anhydride group of the polymer. This step allows the introduction of C–C double bonds at the nanobead surface, which are the moieties that in the next step initiate the NIPAM copolymerization at the bead surface.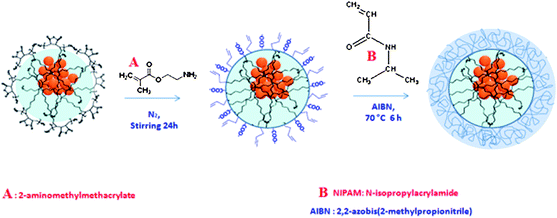 | ||
Scheme 1 To grow the PNIPAM shell on magnetic nanobeads (NBs) starting from polymer coated NBs which expose at their surface anhydride groups, a two-step approach has been applied. In order to introduce C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds groups at the NB surface, the NBs are first primed with 2-aminomethylmethacrylate and in a second step the NIPAM monomers are copolymerized by radical reaction at the bead surface. The structures are just cartoons which underline the functional groups at surface and do not correspond to the real molecular structures. C double bonds groups at the NB surface, the NBs are first primed with 2-aminomethylmethacrylate and in a second step the NIPAM monomers are copolymerized by radical reaction at the bead surface. The structures are just cartoons which underline the functional groups at surface and do not correspond to the real molecular structures. | ||
More in detail, the addition of base (TEA) first deprotonated the AEM, which was subsequently added to the NBs in acetonitrile (ACN) under nitrogen atmosphere. After 24 hours, the modified NBs were precipitated by addition of petroleum ether, and re-dispersed in acetone. The polymerization of NIPAM on the surface of NBs was carried out in water–acetone mixture. To the AEM–NBs in acetone, the radical initiator (AIBN in acetone) was first added, followed by addition of deionized water and NIPAM monomers and the mixture stirred for 6 hours at 70 °C under nitrogen atmosphere.
At the end of the reaction the PNIPAM coated beads were at first magnetically separated and the excess of the NIPAM monomers was removed by few additional precipitation steps which allowed us to completely purify the PNIPAM–NBs from the NIPAM monomers left in the supernatant.
As shown in Fig. 1, while the starting NBs have an average diameter (determined by TEM) of 65 ± 15 nm, the final PNIPAM–NBs have a corresponding diameter of 135 ± 22 nm, and the PNIPAM–NBs samples (as well as the starting NBs) are well dispersed on the TEM grid giving a clear indication of the growth of the PNIPAM shell around each individual starting bead. The change in size due to the polymer growth is also confirmed by the change in hydrodynamic diameter of the PNIPAM–NBs (286 nm) with respect to the diameter of the starting NBs (146 nm) in PBS as measured by DLS (Fig. S2†). (It is also worth to know that even for the intermediate product, the AEM-modified NBs, the TEM picture does not show any aggregation (data not shown) and the DLS diameter in acetone was of 189 nm.)
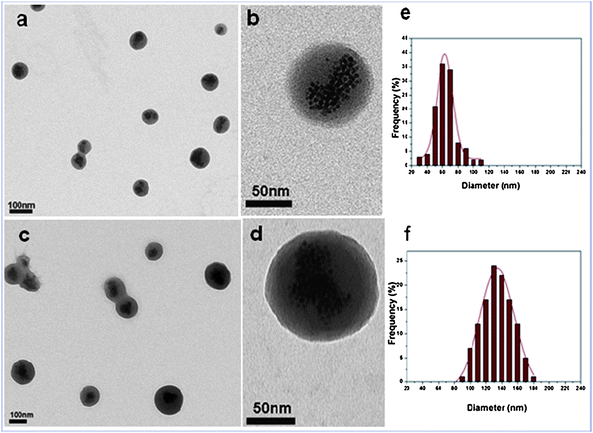 | ||
| Fig. 1 TEM characterization of the starting NBs (a and b) and corresponding PNIPAM–NBs (c and d) at low field and at high magnification. The statistical TEM-diameter measured (f) for the PNIPAM–NBs corresponds to 135 ± 22 nm and (e) for the starting NBs corresponds to 65 ± 15 nm. The statistics have been done on an average of 150 beads measured. | ||
The thermo-responsive properties of the nanobeads before and after the PNIPAM coating provide evidences of the modification of the bead surface. Fig. 2 displays the hydrodynamic diameter of the nanobeads when the temperature was increased from 20 to 55 °C (the heating curve) and vice versa when it was decreased from 55 to 20 °C (the cooling curve) for the starting NBs (Fig. 2b) and for the PNIPAM–NBs in water (Fig. 2a), respectively.
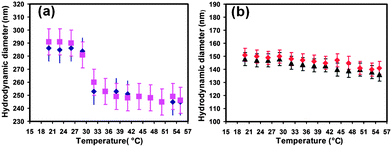 | ||
| Fig. 2 Temperature dependence of hydrodynamic diameter for (a) PNIPAM–NBs (blue diamonds: temperature increased from 20 to 55 °C and violet squares: temperature decreased from 55 to 20 °C) and (b) starting NBs (black triangles: temperature increased from 20 to 55 °C and red diamonds: temperature decreased from 55 to 20 °C). | ||
The selected range of temperature allowed us to study the hydrodynamic size of the NBs above and below the LCST of the PNIPAM, which is, as reported for the linear PNIPAM polymer, around 32 °C. As shown in Fig. 2a, for the PNIPAM–NBs we found a well-defined volume change observed at a temperature around 32 °C. The DLS diameter of the PNIPAM–NBs changes substantially from 286 nm at 20 °C to 250 nm at 55 °C, due to the transition from the random coil to the collapse state of the PNIPAM chains when the temperature was increased above 32 °C. The reversibility of the transition was also observed, as indeed the heating and the cooling curves were found to be basically equal. In the case of starting NBs in water, the hydrodynamic diameter was almost constant at a diameter around 146 nm over the temperature range from 21 to 43 °C and no transition occurs at 32 °C (Fig. 2b) while only a small variation of the DLS size of few nanometres was observed at temperature of 45 °C, which might be due to a slight contraction of the polymer PC18 of which the nanobeads are made occurring at this temperature. The same behavior was observed for the starting NBs in ACN, the solvent in which the nanobeads are prepared (in this case the mean diameter was averaged around 125 nm, data not shown).
The temperature responsive results observed on our PNIPAM–NBs are in accordance with the findings of other groups that have grown the PNIPAM on other types of meso–micro templates (like silica beads or polymer beads).28–31 It is important to point out that on meso-scale objects (100–200 nm), like our PNIPAM–NBs, the polymer contraction involves a change of the volume at individual PNIPAM–NB and it does not involve inter-particle agglomeration. This behavior is obviously different from that of individual PNIPAM-coated IONPs in which the thermo-responsive behavior drives the inter-particle aggregation, leading to micrometre aggregates. The thermo-response behavior of our PNIPAM–NBs is definitely more advantageous when the system is designed for drug delivery as in vivo aggregation should always be avoided.
The encapsulation of NBs within the PNIPAM shell has been additionally confirmed by the FT-IR fingerprint spectra obtained when comparing the starting NBs, the AEM–NBs and the PNIPAM–NBs respectively (Fig. 3). In the IR spectrum of the starting NBs, the peaks at 1780 cm−1 and 1755 cm−1 could be assigned to the CO stretching (asymmetric and symmetric) of the carbonyl groups of the anhydride. After modification with AEM, the carbonyl peak of the carboxylate (per each AEM reacted towards to an anhydride group one carboxylate is produced) was slightly shifted to 1723 cm−1. In addition, due to the formation of amide group, a small peak at 1645 cm−1, likely due to the stretching of amide I carbonyl group (CO/N–H) and another at 1564 cm−1, due to formation of amide II (N–H bend/C–H stretch) were observed. After NIPAM polymerization at the bead surface the peaks at 1645 and 1564 cm−1 were shifted at 1652 and 1544 cm−1 still due to the carbonyl of the amide groups of PNIPAM. Also, the signal of the amide carbonyl peak at 1652 cm−1 after PNIPAM polymerization was much stronger than the AEM-modified NBs, since the ratio of amide groups per bead increased. On the contrary, the peak at 1723 cm−1 of the carbonyl of the carboxylate was much weaker as it is screened by the PNIPAM polymer shell.
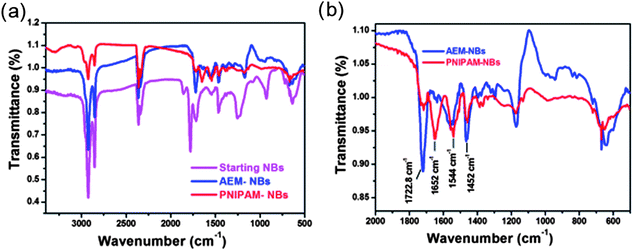 | ||
| Fig. 3 (a) IR-spectra of starting NBs, AEM-modified NBs and PNIPAM–NBs. (b) Detailed IR-spectra of the region of interest for the PNIPAM growth for the AEM-modified beads and PNIPAM–NBs. | ||
Taken all together, the TEM data, the curves DLS diameter versus temperature and the FT-IR spectra provide clear evidence for the surface modification and the PNIPAM polymerization on the surface of the NBs.
Loading of doxorubicin within PNIPAM–NBs and corresponding release experiments
The thermo-responsive change in volume of the polymer shell can be exploited for both the loading and the release of drug molecules. At temperature of 20 °C the PNIPAM–NBs have a larger DLS diameter as the PMIPAM shell is present in a random coil. We exploit the enhanced permeability of the PNIPAM shell at 20 °C to load the doxorubicin (DOXO): the amount of DOXO loaded inside the PNIPAM–NBs has been determined by the difference between a solution of DOXO at initial concentration and that of DOXO left in solution after having magnetically separated the PNIPAM–NBs as sketched in Fig. 4A and B (more details are available in the experimental parts). Thanks to the cumulative effect of several superparamagnetic nanoparticles clustered per beads, the DOXO-loaded PNIPAM–NBs were easily accumulated to the magnet within 1 hour, making the separation protocol much easier and faster without the need of filtering or column purification protocols. In most of the loading experiments starting from a PNIPAM–NBs solution at Fe 50 µg mL−1 concentration, a DOXO solution of 67 µg mL−1 was added and in these conditions we could load within the PNIPAM shell a DOXO amount equal to 50.5 ± 3.5 µg mL−1 (calculated on the average of 7 independent experiments), which corresponds to a loading efficiency of about 75% (actually the highest loading efficiency we could reach). The release profiles have been recorded by using these loading conditions. However, by tuning the initial amount of PNIPAM–NBs and DOXO mixed together, we could vary the loading efficiency. For instance, we kept constant the initial amount of Fe at 12.5 µg mL−1 and by adding an initial amount of DOXO of respectively 10, 16.8, or 100 µg mL−1 we could load within the PNIPAM–NBs respectively 6.5, 12.5 or 24 µg mL−1, which correspond to a loading efficiency respectively of 65, 74, and 25%. (Those sample have been tested later on the tumor cells, however, see Table S1† for the summary of the DOXO loading results obtained.) The loading of DOXO within the PNIPAM shell is also accompanied by an increase in the DLS diameters of the nanobeads from 286 nm for the PNIPAM–NBs to 310 nm for the DOXO-loaded PNIPAM–NBs, while no aggregations were shown on TEM image (Fig. S3†).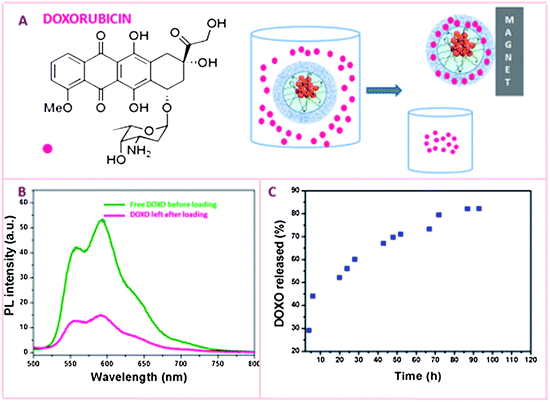 | ||
| Fig. 4 (A) Chemical structure of Doxorubicin and cartoon sketching the loading of DOXO: after mixing the PNIPAM–NBs with DOXO at 20 °C for 12 hours, the DOXO-loaded PNIPAM–NBs are magnetically separated from the free DOXO left in solution. (B) The amount of DOXO loaded is measured by the difference in PL between a solution of DOXO at the same concentration of that mixed with the PNIPAM–NBs and the DOXO left in solution after having magnetically removed the beads. Fluorescence spectra of free DOXO (olive) and DOXO left (pink) in solution after loading experiment. (C) DOXO release kinetics in PBS at 37 °C for the DOXO loaded PNIPAM beads. | ||
The release profile of DOXO from the PNIPAM–NBs was studied at 37 °C in a PBS buffer solution at pH 7.4 as for all the subsequent cell studies the cell cultures were kept at this temperature.
At temperature above the LCST, the PNIPAM shell becomes hydrophobic and leads to the collapse of the polymer layer and consequently allows the water soluble DOXO to diffuse out quickly from the polymer shell. We observed that after 6 hours almost 45% of the loaded DOXO was released, which reached 50% release after 24 hours and reached a percentage of DOXO release of 80% at least 72 hours after keeping the system at 37 °C. The maximum DOXO release percentage reached was around 85% after 110 hours at 37 °C. On the other hand, when keeping the DOXO-loaded PNIPAM–NBs sample at 4 °C for about 72 hours, only 4% of the DOXO encapsulated was released in solution. At 4 °C, after 12 and 24 hours the amount of DOXO released was negligible.
Drug release experiments from DOXO-loaded PNIPAM–NBs on tumor cell lineage
To assess the drug delivery potential of the DOXO-loaded PNIPAM–NBs and the anticancer activity of the released DOXO, in vitro cytotoxicity assays were performed. Tumor KB cells were incubated with the DOXO-loaded carriers at different time courses (24, 48 and 72 h) and at different concentrations of Fe/DOXO administered (1.25/1.25, 2.5/2.5 and 5/5 µg mL−1) (Fig. 5). The experiments were carried out both without (Fig. 5a) and with (Fig. 5b) the application of a small magnet (0.3 T) placed beneath the cell plates and the toxicity behavior was measured by a cell viability MTT assay for respectively, starting NBs, empty PNIPAM–NBs and DOXO-loaded PNIPAM–NBs and was compared to that of free DOXO administered directly to the cell media. In the absence of a magnet (Fig. 5a) it is evident that for the different time points tested and for most of the concentrations of the NBs employed, both the starting NBs and the PNIPAM–NBs showed cell mortality below 20%, which provides an indication for the low toxicity profile of the carriers themselves (actually in those cases, the cells after showing an acute toxicity at 24 h, had the time to recover after 48 and 72 h). Those findings are in accordance with the data reported by Vihola et al. on bare PNIPAM polymer molecules tested on two different cell lines; a negligible toxicity of the PNIPAM polymer itself has been shown in their study.32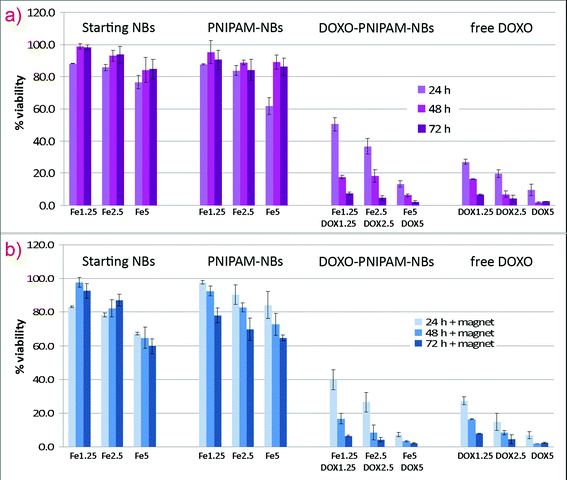 | ||
| Fig. 5 In vitro cytotoxicity assay of PNIPAM–NBs loaded with DOXO performed (a) without and (b) with the application of an external magnet. KB cells were incubated for 24, 48 and 72 h at 37 °C with the loaded systems at different amounts of beads (1.25, 2.5 and 5 µg mL−1 of Fe) and DOXO (1.25, 2.5 and 5 µg mL−1). The cytotoxicity of the starting beads, of the empty PNIPAM–NBs and of the free DOXO has been also tested and compared to that of DOXO-loaded PNIPAM–NBs. | ||
More significant is the comparison of the toxicity observed at 24 hours between the DOXO-loaded PNIPAM–NBs and that of free DOXO administered to the cells for the three concentrations tested. The encapsulation of the DOXO within the PNIPAM shell retards the toxic effect of DOXO on the cells as observed from the comparison of cell viability at the time point of 24 hours (for instance for Fe/DOXO 1.25/1.25 it corresponds to a cell survival of respectively 50% for the DOXO-loaded PNIPAM–NBs against the 25% of the free DOXO, and the trend is the same also for the other Fe/DOXO ratios). On the contrary, the long term toxicity, measured at the time points of 48 and 72 hours, for the DOXO-loaded PNIPAM–NBs tend to reach a cell viability value that is comparable with that of free DOXO, confirming the continuous time release of DOXO from the PNIPAM shell on longer time scale.
It should be noticed that the DOXO release profile on the system itself, reported in Fig. 4, well accounts for the trend observed on the cells, since the curve almost reached the release saturation when the sample was kept at 37 °C for a period longer than 3 days, while after 24 h only 50% of DOXO was released.
Importantly, the presence of a static magnet placed beneath the Petri dish should trigger the intracellular uptake of the beads due to the magnetic attraction of the magnetic NBs towards the magnet, thus favoring the amount of beads in contact with the cell membrane and consequently up-taken by the cells.27 Indeed, this trend is confirmed by the experimental data (Fig. 5b); the presence of the magnet induces an appreciable increase of the cytotoxicity compared to the corresponding samples incubated without magnet for all the Fe/DOXO concentrations tested. For instance, the viability of cells doped with DOXO-loaded PNIPAM beads at Fe/DOXO concentration equal to 1.25 µg mL−1 after 24 h is 40% when the incubation occurred with the magnet, while it corresponds to 50% without the magnet. One should be aware that in those experiments, it is not possible to discriminate the toxicity due to the DOXO release from extracellular nanobeads (even the DOXO released from non-internalized carriers in the cell medium could cross the cell membrane and enter the nuclei, as well). Such effect, however, should occur in the same manner in both samples with and without the magnet (to discriminate such effect a different thermo-responsive polymer with a shrinking temperature over 37 °C is required). The magnetic contribution to the cytotoxicity enhancement is more pronounced for short incubation times (at 24 h), while after longer time, at 48 and 72 h, the DOXO release effect prevails more than the accumulation effect (an amount of DOXO enough to kill the tumor cells has been released even without the application of the magnet).
To test the effect on the cell mortality when different amounts of DOXO have been encapsulated within the beads, KB cells were administered with PNIPAM–NBs (Fe 1.25 µg mL−1, this nanobead concentration corresponds to the most negligible toxicity of the system itself) loaded with different amounts of DOXO, respectively 0.65, 1.25 and 2.5 µg mL−1. (Fig. 6). As expected, lower DOXO concentration corresponds to higher viability at 24 h, although after 72 h more than 80% cells were found dead. On the other hand, when the cells were incubated with 2.5 µg mL−1DOXO, after 24 h around 50% of cytotoxicity was recorded. These data also imply that a very low amount of drug (less than 1 µg mL−1) is needed to exert a high cytotoxic effect, on a longer time scale of 72 h. All these features could make this carrier system a powerful tool for in vivo chemotherapy against cancer, where it is fundamental that the drug is active not immediately, after the intra body injection, but only after the tumor site has been reached.33,34 Also, lower drug amount needs to be administered to get complete tumor regression while minimizing the risk to develop chemoresistance.35–37
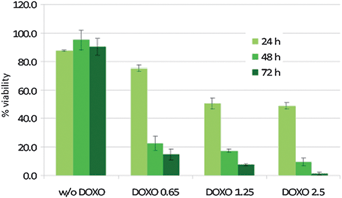 | ||
| Fig. 6 Comparative cell viability assay of KB cells incubated with DOXO-loaded PNIPAM–NBs by keeping constant the amount of NBs (1.25 µg mL−1Fe) and increasing the amount of DOXO loaded within the PNIPAM shell (respectively, 0.65, 1.25 and 2.5 µg mL−1). The cytotoxicity of the empty PNIPAM beads is also reported. For each sample the toxicity has been measured after an incubation time of respectively 24, 48 and 72 hours, at 37 °C. | ||
Although it is not straightforward to compare our results with those obtained by other groups (as indeed different groups have used different ways to estimate the amount of iron oxide based nanostructures administered to the cells in their studies,25,38) it is remarkable to mention that in our experiments we deliver a DOXO concentration in the same range used by other groups,38–41 however, by employing an amount of nanostructures (measured in terms of iron), lower with respect to that of other groups41,42 (to give an example the highest amount of iron of 5 µg mL−1 used in this work corresponds to an amount of iron oxide nanoparticles of 6 nm in diameter of about 20 nM).
In order to correlate the toxicity data with the up-taking of the nanobeads, the intracellular amount of PNIPAM–NBs and starting NBs in terms of intracellular Fe amount was measured by means of elemental analysis (Fig. 7 and Table S2, ESI†).
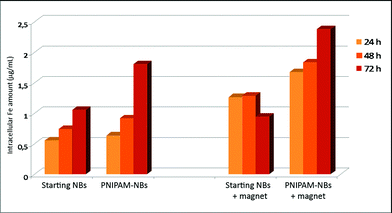 | ||
Fig. 7
Intracellular amount of Fe content (µg mL−1) for cells treated with starting beads and PNIPAM coated beads at an extracellular concentration of 2.5 µg mL−1. 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 KB cells were incubated for 24, 48 and 72 hours at 37 °C with and without the application of an external magnet. After the incubation time the cells were digested and the Fe content was determined by elemental analysis. 000 KB cells were incubated for 24, 48 and 72 hours at 37 °C with and without the application of an external magnet. After the incubation time the cells were digested and the Fe content was determined by elemental analysis. | ||
It is clear that either with or without the magnet the PNIPAM–NBs were internalized by the KB cells more efficiently than the starting NBs (Fig. 7). Since the surface of both samples lacks any cellular addressing ligand or recognition molecule that could promote the interaction with the cell membrane and the internalization, this behavior can be attributed to the different surface charge and structure displayed by the two samples: according to the ζ-potential measurements, PNIPAM–NBs are less negatively charged compared to the starting beads (−10 mV and −40 mV, respectively, in PBS) and indeed the reduced repulsion at the negative cell membranes facilitates the uptaking.43 In the case of the PNIPAM–NBs some of the carboxyl moieties present at the starting NB surface are used in the reaction with the AEM priming molecules for the PNIPAM growth and thus do not contribute to the final negative surface charge of the carriers. Additionally, as also mentioned by others when PNIPAM–gold nanostructures have been studied,44 the up-taking might be driven by the hydrophobic interactions between the cell membrane and the PNIPAM polymer. At 37 °C, i.e. over the LCST, the PNIPAM polymer is in the collapsed and hydrophobic conformation; indeed the interaction with the phospholipid part of the membrane could preferentially occur, thus enhancing the PNIPAM–NBs up-taking with respect to the starting NBs.
In addition the higher cytotoxicity of the PNIPAM–NBs with respect to the starting NBs, is confirmed by the observation that the presence of the magnet considerably increases the internalization of the PNIPAM–NBs: after 72 hours the intracellular Fe amount found was 2.5 µg mL−1, equivalent to the amount of PNIPAM–NBs externally administered to the cells. We conclude that the less negative surface provided by the PNIPAM shell to the beads, together with the magnetic manipulation of the inner core of the PNIPAM–NB system account for the efficient up-taking of the system developed in this work: after 72 hours nearly 100% of the PNIPAM–NBs administered to the cells was taken up by the cells, when the magnet was placed beneath the cell dish (Fig. 7). In Fig. 7 one should also notice that in general for all the samples the up-taking increases with time. This trend, however, is not maintained for the starting NBs after 72 hours in the presence of the magnet. This behavior might be attributed to the reduced stability of the starting NBs that tend to aggregate and stick to the cell plate due to the magnetic attraction resulting in a lower number of beads present in solution.
The cellular internalization of both starting NBs and PNIPAM–NBs was also visually monitored by TEM characterization of cells incubated with both starting NBs and PNIPAM–NBs samples. Fig. 8 shows the intracellular localization of the starting beads after 24 h incubation. Early and late endosomes (Fig. 8b) were present in the same cell: the early endosomes were quite large, displayed an irregular shape and contained a considerable amount of beads. On the other hand, late endosome and endolysosomal compartments were evident even after 48 h (Fig. S5†): single iron oxide nanoparticles were held inside the lysosomes suggesting that the pH conditions of the lysosomes could degrade the polymer layer. However, further studies on longer time scale need to be carried out in order to explain these findings.
 | ||
| Fig. 8 TEM images of KB cells incubated with the starting NBs (a) after 24 h at 37 °C. (b) Higher magnification of a portion of the same image showing an endosome containing a considerable amount of nanobeads, many nanostructures having an inner dark core corresponding to IONPs and a lighter polymer shell are well distinguishable within the endosome. | ||
Even PNIPAM–NBs were phagocyted and retained into the endosomal compartments after 24 h (Fig. 9a). Fig. 9b shows the last stage of the membrane invagination during the formation of an early endosome. Even from the structural morphology of the beads uptaken within the cells it is possible to recognize the PNIPAM–NBs as indeed in this case the polymer layer is much thicker with respect to the starting NBs. Also in this case, from a qualitative point of view, after 48 h the cells were enriched with PNIPAM vesicles (Fig. S6†) and after 3 days the cells appeared overflowing (Fig. S6†). Those TEM images, tough qualitatively, confirm the above reported trend of preferential up-taking by the cells of the PNIPAM–NBs with respect to the starting NBs (Fig. S6†).
 | ||
| Fig. 9 TEM images of KB cells incubated with the PNIPAM beads after 24 h at 37 °C. Low magnification image showing the PNIPAM–NBs up-taken by the cell and accumulated in the endosome (see the inset). (b) Invagination of the cell membrane and formation of the endosome containing the PNIPAM–NBs. | ||
KB cells once incubated with DOXO-loaded PNIPAM–NBs (2.5/2.5 µg mL−1Fe/DOXO concentration) were imaged at different time delays of 2, 7 and 15 h incubation (Fig. 10a–c). The fluorescent signal of DOXO allowed us to follow the time dependent DOXO internalization. After 2 h, a red fluorescent signal of DOXO was recorded on cells, which significantly increased with time despite the number of cells in the recorded area after 15 h was less than that recorded after 7 h (most likely because the cells killed by the DOXO release were detached from the substrate). These data were also confirmed by looking at fixed cells after 15 h. In Fig. 10d–f it is possible to distinguish between the necrotic cells and those that had up-taken DOXO without being killed: the blue fluorescent signal of nuclei staining with DAPI for the necrotic cells (Fig. 10d) is merged with the red fluorescent signal of DOXO (Fig. 10e) resulting in a final violet color in the merged images (Fig. 10d).
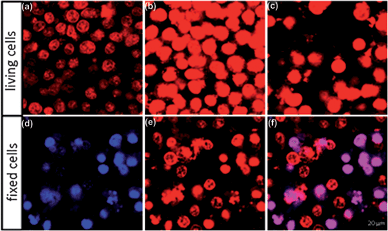 | ||
| Fig. 10 (a–c) Confocal images of living KB cells exposed to PNIPAM–NBs loaded with DOXO. Images were acquired after 2 (a), 7 (b) and 15 h (c) incubation time. (d–f) Confocal images of fixed cells treated with PNIPAM–NBs. The DAPI stains in blue the nuclei, while the red signal visualizes the DOXO (e) and the merged channel (f) allows the better visualization of dead cells, the violet spots. | ||
Conclusion
In this work, starting from magnetic nanobeads consisting of multiple iron oxide nanoparticles clustered together through a polymer, we have added a thermo-sensitive shell based on PNIPAM and we have exploited the PNIPAM–NBs as carriers for the doxorubicin.The nanostructures here developed as drug carrier represent a second generation of multifunctional nano-materials based on multiple iron oxide nanoparticles which combine the benefit to have a stronger magnetic core, and thus a stronger response to the external magnet, with the advantage to keep the super-paramagnetic behavior of the nanoparticles and thus easily to be dispersed in solution whenever the magnet is removed. In addition, the DOXO encapsulation within the PNIPAM shell allows to protect the external environment (the healthy tissues) from the toxic drug and also to slowly release the embedded drug. The DOXO released by the nanostructure exhibits pharmacological activity on tumor cells, and the drug release profile is clearly temperature and time dependent. Also, in concomitance to the physical force exerted by the static magnet by properly adjusting the incubation time, and the surface charge of the carrier nearly 100% cellular up-taking efficiency was achieved.
All these performances have been achieved in a miniaturized nanostructure, which has a final diameter still below the critical threshold (below 200 nm) for applications in vivo and with a control on the overall morphology and thus functionality.
While in this work we provide the proof of concept for the use of magnetic beads decorated by PNIPAM as thermo-responsive polymer with a LCST of about 32 °C, we could envision that by appropriate selection of the comonomer units (such as for instance a combination of NIPAM-co-acrylic acid-co-ethyl methacrylate),45–47 the transition temperature of the polymer could be switched to higher temperature (around 40–43 °C). The resulting thermo-responsive polymer magnetic nanobeads, could be used as carrier to release the drug at temperature higher than 37 °C, the physiological body temperature. The heat produced by the magnetic nanoparticles under appropriate alternating magnetic field might be used to damage the cancer cells by hyperthermia and at the same time to trigger the drug release. By using such system the “aspecific” drug release could be prevented. The magnetic heat-ability of MNPs might be exploited not only to induce hyperthermia, but the same thermal effect produced by the magnetic nanoparticles might be exploitable to trigger the conformation transition of the thermo-responsive polymer and thus the subsequent drug release. The work done in this study poses the base for the development of such multifunctional nanostructures.
Materials and methods
Chemicals
N-Isopropylacrylamide (NIPAM, 97%), 2,2′-azobis(2-methylpropionitrile) (AIBN), 2-aminoethyl methacrylate hydrochloride (AEM), triethylamine, (TEA, 99%), poly(maleic anhydride-alt-1-octadecene) Mn 30000–50000 (PC18), and doxorubicin hydrochloride (DOXO) were supplied by Sigma Aldrich and were used without any further purification. Solvents like acetonitrile (ACN, HPLC grade), tetrahydrofuran (THF), petroleum ether, and acetone were supplied by Carlo Erba.
Synthesis of nanobeads (NBs)
To prepare magnetic nanobeads, in a 4 mL glass vial, 50 µL of IONPs (15.4 µM nanoparticle concentration, d = 6 nm) in toluene were dried to remove toluene completely and the nanoparticles were redissolved in 150 µL of THF. Soon after, we added 50 µL of a stock solution of Poly(maleic anhydride-alt-1-octadecene) (PC18) in THF (50 mM, this concentration refers to the polymer monomer units). The mixture was shaken at 1000 rpm at 20 °C for 60 minutes. Subsequently, 800 µL of acetonitrile (ACN) were added at a flow rate of 250 µL min−1. In order to improve size distribution, after the beads were formed, a magnet (0.3 T) was placed to one side of the vial and the first portion attracted to the magnet (within the first 10 min) was discarded. To the recovered supernatant, the magnet was applied to the glass wall for 12 hours, in order to quantitatively recover to the magnet all the beads left in solution. To scale up the beads production, the procedure reported was repeated 10 times and the beads recovered to the magnet, from each batch, were then redissolved in fresh ACN to a concentration of nanobeads corresponding to 655 µg mL−1 of Fe, as determined by elemental analysis. Those starting NBs in ACN are used for the next step of PNIPAM growth. For the comparative behavior of the starting NBs with the PNIPAM–NBs, the magnetic NBs prepared in ACN were simply transferred in water, for the cell study by attracting them to magnet, removing the ACN and adding fresh PBS buffer (pH 7.4). Also in this case, the nanobeads concentration was always referred as Fe content (µg mL−1) and it was determined by elemental analysis.Functionalization of nanobeads with 2-aminoethyl methacrylate (AEM–NBs)
To prime the nanobead surface with 2-aminoethylmethacrylate in a 25 mL three-necked round-bottom flask, to 0.009 g of AEM, 1 mL of a solution of TEA in ACN (2 M) was first added. Then 2 mL of a NBs solution in ACN at 655µg mL−1 of Fe were also added and the reaction volume was adjusted to 15 mL with ACN. The solution was left under stirring for 24 hours at 18 °C in nitrogen atmosphere. Finally, 10 mL of petroleum ether were added to the reaction mixture in order to precipitate the modified NBs, followed by re-dispersion in ACN and magnetic separation (as described above). In order to be sure to remove the excess of AEM the purification step was repeated twice. At the last step the modified NBs were dispersed in acetone at a concentration of 377 µg mL−1 of Fe (as determined by ICP).Polymerization of NIPAM on the surface of NBs (PNIPAM–NBs)
To initiate polymerization of NIPAM at the bead surface, in a three-necked round-bottom flask, 0.006 g of AIBN was dissolved in 10 mL of acetone, and 2 mL of AEM-modified NB solution (377µg mL−1Fe) were added. Then, 0.2 g (1.76 mmol) of NIPAM and 20 mL of water were added. The reaction vessels were connected to a reflux condenser and purged with nitrogen for 30 minutes. The mixture was heated to 70 °C and the reaction was allowed to proceed for 6 hours under nitrogen atmosphere. The final product was collected by magnetic separation and was easily re-dispersed in deionized water. To remove the excess of monomers, the product was centrifuged at 4000 rpm for 30 minutes and the precipitate was redissolved in water. This process was repeated three times.Doxorubicin loading experiment
In order to load DOXO within PNIPAM–NBs 100 µL of a solution of DOXO in water (1 mg mL−1) were added to 1.4 mL of PNIPAM–NBs in water (Fe concentration 50 µg mL−1) and the mixture was shaken overnight in the dark at 20 °C. The solution was then centrifuged at 4000 rpm, and the precipitate was redissolved in water (1.5 mL) and kept near the magnet for 1 additional hour, in order to collect the PNIPAM–NBs at the magnet and discard the free DOXO in solution. This step was repeated twice in order to remove completely the free DOXO in solution.The amount of DOXO loaded inside the beads was determined by photoluminescence measurements, given as the difference between a solution of DOXO at the initial concentration used for the experiment and that of DOXO left in solution (in the supernatant) after the loading time. The DOXO has a maximum of emission at 591 nm when excited at 450 nm. The amount of DOXO left in the supernatant was measured by a calibration PL curve obtained by plotting the PL intensity at 591 nm versus the DOXO concentration of standard solutions. Importantly, as the PL signal of DOXO changes over time, we have always prepared a DOXO solution at the same initial concentration (67µg mL−1) of that added to the PNIPAM–NBs and we kept this solution in the same conditions (overnight at 20 °C in the dark) of those under which are kept the samples. This DOXO solution was used as stock solution for the preparation of all the standards at known concentration.
The drug loading efficiency was calculated as follows:
| Encapsulation efficiency (%) = 100 × ((initial amount of DOXO − amount of DOXO left in the supernatant)/initial amount of DOXO) |
DOXO release experiment from the DOXO-loaded PNIPAM–NBs
We have followed the DOXO release by optical absorption (the DOXO has a maximum peak of adsorption at a wavelength of 483 nm). In a glass vial, 2 mL of DOXO–PNIPAM–NB (50 µg mL−1Fe) solutions in PBS were shaken gently at 37 °C. After a time lapse (data points were collected at different times respectively: 4, 6, 20, 24, 28, 48, 52, 67, 72, 87, 96 and 110 hours), a magnet (0.3 T) was placed to the side wall of the vial and it was kept for 1 hour, the time needed to collect all the beads at the magnet. The supernatant was then recovered and the emission spectrum was recorded on it. The amount of released DOXO was determined by a calibration curve obtained by plotting the DOXO adsorption at 483 nm wavelength versus the DOXO concentration of standard solutions (see ESI, Fig. S1†). After each time step, the DOXO–PNIPAM–NBs re-collected to the magnet were redissolved in 2 mL of fresh PBS buffer and kept for the additional time lapse before being magnetically separated from the supernatant.Cell culture and cell viability assay
KB cells were grown at 37 °C and under 5% CO2 atmosphere in RPMI-1640 medium, supplemented with L-glutamine (2 mM), penicillin (100 units mL−1), streptomycin (100 µg mL−1), and 10% heat-inactivated fetal bovine serum (FBS).A viability assay (MTT test) was performed using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide. In detail, 5 × 104cells suspended in 1 mL of medium were seeded in each well of a 12 well-plate, and after 24 h incubation at 37 °C, the medium was replaced with 1 mL of fresh medium containing the DOXO-loaded PNIPAM–NBs at different Fe/DOXO concentrations (1.25/0.65, 1.25/1.25, 1.25/2.5, 2.5/2.5, 5/5 µg mL−1). Cells were kept at 37 °C for 24, 48 or 72h. Then the medium was removed, the cells were washed three times with PBS, and 1 mL of fresh serum-free medium containing 1 mg mL−1 of MTT was added into each well. After 3 hours of incubation at 37 °C the medium was removed and the precipitated dark crystals were dissolved in 2 mL of DMSO, leading to a violet solution whose absorbance at 570 nm was determined. Each assay was performed in triplicate and with or without the application of a static magnetic field (0.2 T) under the cell plates. The cytotoxicity of the free DOXO, of the unloaded PNIPAM–NBs and of the starting NBs at the same concentrations used for the DOXO experiment was also assayed.
Determination of the intracellular Fe concentration
To estimate the intracellular Fe concentration, 105cells suspended in 2 mL of medium were seeded in each well of a 6 well-plate. After 24 h incubation at 37 °C in each well, the medium was replaced with 2 mL of fresh medium containing either the unloaded PNIPAM–NBs or the starting NBs at a Fe concentration equal to 2.5 µg mL−1. Cells were incubated at 37 °C for 24, 48 or 72 hours with or without the application of the magnetic field. Thereafter, the medium was removed; the cells were washed twice with PBS and trypsinized by addition of 500 µL of trypsin per well. After 5 min incubation at 37 °C, the cell suspension was collected and 1.5 mL of PBS was added in order to recover any remaining cells from each well. The suspension was then centrifuged, the supernatant removed and 2 mL of a concentrated HCl solution was added to digest the cells (the solution was left for 48 hours). The intracellular Fe concentration was measured by means of elemental analysis (ICP-OES spectrometer, iCAP 6500, Thermo).Preparation of cell samples for TEM characterization
4 × 105cells suspended in 8 mL of medium were seeded in a culture dish. After 24 h incubation at 37 °C the medium was replaced with 8 mL of fresh medium containing either the unloaded PNIPAM beads or the starting beads at a Fe concentration equal to 2.5 µg mL−1. Cells were incubated at 37 °C for 24, 48 or 72 h. Then, they were washed with PBS and fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer at 4 °C for 30 min. The fixed specimens were washed three times with the same buffer and 1% osmium tetroxide in cacodylate buffer was added for 1h. Thereafter, the cells were washed again and dehydrated with 25%, 50%, 75% and 100% acetone. Two steps of infiltration in a mixture of resin/acetone (1/1 and 2/1 ratios) followed and finally the specimens were embedded in 100% resin at 60 °C for 48 h. Ultrathin sections (100 nm thick) were cut on an Ultramicrotome (Leika), stained with lead citrate and observed under a JEOL JEM-1011 microscope operating at 100 kV.Instruments used
ζ-Potential and dynamic light measurements (DLS) were performed on a Zetasizer Nano ZS90 (Malvern, USA) equipped with a 4.0 mW He–Ne laser operating at 633nm and an avalanche photodiode detector. Measurements were made at 25 °C in deionized water. All the samples were filtered using 0.45µm filters before analysis. DLS measurements were made at temperatures ranging from 20 to 55 °C at a temperature interval of 3 °C, allowing temperature stabilization for a period of 15 minutes before each measurement. For the experiments, the concentration of NBs samples (the starting NBs and the PNIPAM–NBs) was of 5 µg mL−1Fe. The UV-visible absorption spectra were measured using a Varian Cary 300 UV-Vis spectrophotometer while the Photoluminescence (PL) spectra were recorded on a Cary Eclipse spectrophotometer. The FT-IR measurements were recorded on Bruker vertex V80 FT-IR spectrometer. The TEM images were recorded on a JEOL jem 1011 microscope operated at an accelerating voltage of 100 kV. TEM samples of the bare materials were prepared by dropping a diluted solution of nanobeads in water on carbon-coated copper grids and letting the solvent evaporate. Confocal microscopy images of living KB cells incubated with the DOXO–PNIPAM–NBs were recorded on a Nikon Eclipse Ti microscope equipped with an argon laser source (excitation at 488 nm). Images were recorded at time 0, 2, 7 and 15 h using an acquisition window set at 590 ± 20 nm. For the image of fixed cells, the cells previously incubated for 15 hours at 37 °C with DOXO–PNIPAM–NBs were washed with PBS, fixed with 4% PBS buffered paraformaldehyde, and the nuclei stained with 4′-6-diamidino-2-phenylindole (DAPI). The specimens were observed under the confocal microscope, using two laser excitation sources (at 405 and 488 nm), and two acquisition channels at 450 ± 20 nm and 590 ± 20 nm respectively, for DAPI and DOXO.Acknowledgements
This work was supported in part by the European project Magnifyco (Contract NMP4-SL-2009-228622). We thank Mario Malerba for TEM sample preparation and Dr Liberato Manna for helpful discussion.References
- M. Das, H. Zhang and E. Kumacheva, Annu. Rev. Mater. Res., 2006, 36, 117–142 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- A. Chilkoti, M. R. Dreher, D. E. Meyer and D. Raucher, Adv. Drug Delivery Rev., 2002, 54, 613–630 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- T. Y. Liu, S. H. Hu, D. M. Liu, S. Y. Chen and I. W. Chen, Nano Today, 2009, 4, 52–65 CrossRef CAS.
- A. Figuerola, R. Di Corato, L. Manna and T. Pellegrino, Pharmacol. Res., 2010, 62, 126–143 CrossRef CAS.
- F. Gazeau, M. Levy and C. Wilhelm, Nanomedicine, 2008, 3, 831–844 CrossRef CAS.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS.
- S. R. Deka, A. Quarta, R. Di Corato, A. Falqui, L. Manna, R. Cingolani and T. Pellegrino, Langmuir, 2010, 26, 10315–10324 CrossRef CAS.
- A. M. Schmidt, Colloid Polym. Sci., 2007, 285, 953–966 CrossRef CAS.
- Y. H. Lien and T. M. Wu, J. Colloid Interface Sci., 2008, 326, 517–521 CrossRef CAS.
- N. S. Satarkar, D. Biswal and J. Z. Hilt, Soft Matter, 2010, 6, 2364–2371 RSC.
- A. Sánchez-Iglesias, M. Grzelczak, B. Rodríguez-Gonzaález, P. Guardia-Girós, I. Pastoriza-Santos, J. Pérez-Juste, M. Prato and L. M. Liz-Marzán, ACS Nano, 2009, 3, 3184–3190 CrossRef CAS.
- J. Rubio-Retama, N. E. Zafeiropoulos, C. Serafinelli, R. Rojas-Reyna, B. Voit, E. Lopez Cabarcos and M. Stamm, Langmuir, 2007, 23, 10280–10285 CrossRef CAS.
- L. B. Chen, F. Zhang and C. C. Wang, Small, 2009, 5, 621–628 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503–7509 CrossRef CAS.
- A. P. Herrera, M. Rodriguez, M. Torres-Lugo and C. Rinaldi, J. Mater. Chem., 2008, 18, 855–858 RSC.
- J. Qin, Y. S. Jo and M. Muhammed, Angew. Chem., Int. Ed., 2009, 48, 7845–7849 CrossRef CAS.
- D. L. LesliePelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770–1783 CrossRef CAS.
- A. G. Roca, M. P. Morales, K. O'Grady and C. J. Serna, Nanotechnology, 2006, 17, 2783–2788 CrossRef CAS.
- Y. B. Sun, X. B. Ding, Z. H. Zheng, X. Cheng, X. H. Hu and Y. X. Peng, Chem. Commun., 2006, 2765–2767 RSC.
- M. Rahimi, A. Wadajakar, K. Subramanian, M. Yousef, W. Cui, J. T. Hsieh and K. T. Nguyen, Nanomedicine, 2010, 6, 672–680 CAS.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22 CrossRef.
- R. Di Corato, P. Piacenza, M. Musaro, R. Buonsanti, P. D. Cozzoli, M. Zambianchi, G. Barbarella, R. Cingolani, L. Manna and T. Pellegrino, Macromol. Biosci., 2009, 9, 952–958 CrossRef CAS.
- D. Suzuki and H. Kawaguchi, Colloid Polym. Sci., 2006, 284, 1443–1451 CrossRef CAS.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS.
- M. Ballauff and Y. Lu, Polymer, 2007, 48, 1815–1823 CrossRef CAS.
- R. Contreras-Caceres, A. Sanchez-Iglesias, M. Karg, I. Pastoriza-Santos, J. Perez-Juste, J. Pacifico, T. Hellweg, A. Fernandez-Barbero and L. M. Liz-Marzan, Adv. Mater., 2008, 20, 1666–1670 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- L. Wang and J. Q. Sun, J. Mater. Chem., 2008, 18, 4042–4049 RSC.
- T. Minko, S. S. Dharap, R. I. Pakunlu and Y. Wang, Curr. Drug Targets, 2004, 5, 389–406 Search PubMed.
- G. Szakacs, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef CAS.
- M. Susa, A. K. Iyer, K. Ryu, F. J. Hornicek, H. Mankin, M. M. Amiji and Z. F. Duan, BMC Cancer, 2009, 9, 12 CrossRef.
- E. Munnier, S. Cohen-Jonathan, C. Linassier, L. Douziech-Eyrolles, H. Marchais, M. Souce, K. Herve, P. Dubois and I. Chourpa, Int. J. Pharm., 2008, 363, 170–176 CrossRef CAS.
- J. W. Liu, Y. Zhang, C. Y. Wang, R. Z. Xu, Z. P. Chen and N. Gu, J. Phys. Chem. C, 2010, 114, 7673–7679 CrossRef CAS.
- J. S. Guthi, S. G. Yang, G. Huang, S. Z. Li, C. Khemtong, C. W. Kessinger, M. Peyton, J. D. Minna, K. C. Brown and J. M. Gao, Mol. Pharmaceutics, 2010, 7, 32–40 CrossRef CAS.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552–557 CrossRef CAS.
- G. B. Hong, R. X. Yuan, B. L. Liang, J. Shen, X. Q. Yang and X. T. Shuai, Biomed. Microdevices, 2008, 10, 693–700 CrossRef CAS.
- R. R. Arvizo, O. R. Miranda, M. A. Thompson, C. M. Pabelick, R. Bhattacharya, J. D. Robertson, V. M. Rotello, Y. S. Prakash and P. Mukherjee, Nano Lett., 2010, 10, 2543–2548 CrossRef CAS.
- M. Liang, I. C. Lin, M. R. Whittaker, R. F. Minchin, M. J. Monteiro and I. Toth, ACS Nano, 2010, 4, 403–413 CrossRef CAS.
- M. J. Tiera, G. R. dos Santos, V. A. D. Tiera, N. A. B. Vieira, E. Frolini, R. C. da Silva and W. Loh, Colloid Polym. Sci., 2005, 283, 662–670 CrossRef CAS.
- A. Zintchenko, M. Ogris and E. Wagner, Bioconjugate Chem., 2006, 17, 766–772 CrossRef CAS.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: A table summarizing the DOXO loading parameters, DLS of some steps of the reaction and additional TEM characterization images of the DOXO loaded nanobeads, and additional TEM characterization of cells treated with the nanobeads. See DOI: 10.1039/c0nr00570c |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |