Nanoparticles for cell labeling
Ashwinkumar
Bhirde
,
Jin
Xie
,
Maggie
Swierczewska
and
Xiaoyuan
Chen
*
Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institute of Health (NIH), Bethesda, MD 20892, USA. E-mail: shawn.chen@nih.gov
First published on 11th October 2010
Abstract
Cell based therapeutics are emerging as powerful regimens. To better understand the migration and proliferation mechanisms of implanted cells, a means to track cells in living subjects is essential, and to achieve that, a number of cell labeling techniques have been developed. Nanoparticles, with their superior physical properties, have become the materials of choice in many investigations along this line. Owing to inherent magnetic, optical or acoustic attributes, these nanoparticles can be detected by corresponding imaging modalities in living subjects at a high spatial and temporal resolution. These features allow implanted cells to be separated from host cells; and have advantages over traditional histological methods, as they permit non-invasive, real-time tracking in vivo. This review attempts to give a summary of progress in using nanotechnology to monitor cell trafficking. We will focus on direct cell labeling techniques, in which cells ingest nanoparticles that bear traceable signals, such as iron oxide or quantum dots. Ferritin and MagA reporter genes that can package endogenous iron or iron supplement into iron oxide nanoparticles will also be discussed.
 Ashwinkumar Bhirde | Ashwinkumar Bhirde obtained his B.E. in chemical engineering in 1999 from Karnataka University in India. He then received MS in chemical engineering in 2004 from Lamar University Texas under Professor Shyam Shukla. Later he obtained his Ph.D. in chemistry under Professor James Rusling at the University of Connecticut in 2010 in collaboration with Drs. Silvio Gutkind and Vyomesh Patel from NIDCR/NIH. In 2010 he joined Dr Xiaoyuan Chen's group at the NIBIB/NIH where he has since been a member of the nanomedicine group. His current research is focused on development of nanomaterial based imaging and drug delivery systems for biomedical applications. |
 Jin Xie | Jin Xie received his B.S. in chemistry in 2003 from Nanjing University, China. In 2004 he joined a Ph.D. program in the Department of Chemistry at Brown University under the supervision of Prof. Shouheng Sun. His Ph.D. work involved synthesis of high moment magnetic nanoparticles. After obtaining his Ph.D. he joined Dr Xiaoyuan Chen's group in the Molecular Imaging Program at Stanford (MIPS), and in 2009, he moved to the National Institute of Biomedical Imaging and Bioengineering (NIBIB) where he has since been a member of the Laboratory for Molecular Imaging and Nanomedicine (LOMIN). His current research is focused on development of nanoparticle-based theranostics and activatable probes. |
 Maggie Swierczewska | Magdalena Swierczewska is a graduate student in the Biomedical Engineering Department at Stony Brook University working towards her Ph.D thesis at Dr Chen's Laboratory of Molecular Imaging and Nanomedicine of the NIBIB/NIH. Her previous works include the development and characterization of novel nanomaterials such as single-walled carbon nanotubes and polypeptides. Using her background in nanotechnology and material science, Maggie is focused on the development of novel nanoplatforms for ultrasensitive diagnostics. |
 Xiaoyuan Chen | Xiaoyuan (Shawn) Chen was born in Jiangsu, China in 1974. Dr Chen received his Ph.D. in chemistry from the University of Idaho in 1999. After two quick postdocs at Syracuse University and Washington University in St. Louis, he joined the University of Southern California as an Assistant Professor of Radiology. He then moved to Stanford University in 2004. In 2009, he joined the intramural research program of the National Institute of Biomedical Imaging and Bioengineering (NIBIB) as a tenured senior investigator and Chief, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), specializing in synthesizing molecular imaging probes for multimodality molecular imaging as well as high sensitivity nanosensors. |
1. Introduction
Cell transplantation is an emerging therapeutic avenue with potential applications on many fronts.1 In particular, increasing attention has been paid to stem cell and progenitor cell based research for its potential in regenerative medicine, such as in central nervous system (CNS)-related diseases and myocardial regeneration.2 Challenges in the related investigation are to distinguish the implanted cells from the host tissue cells and to monitor their survival, migration, differentiation and regenerative impact in living subjects.3 To meet those challenges, ex vivocell labeling techniques have been generated, which, when combined with state-of-the-art imaging modalities, provide a means of tracking the cell implants in a non-invasive and real-time fashion over a reasonably long observation period.There are direct and indirect ways of labeling cells with nanoparticles. Direct cell labeling refers to the strategy of loading cells with probes before engraftment into the host. A number of nanoparticles with unique physical properties have found great use in such applications. Possessing a size of several to hundred nanometers, these nanoparticles can be taken up by cells before implantation serve as dyes to facilitate tracking of cells. Iron oxide nanoparticles, for instance, have long been used as magnetic resonance imaging (MRI) contrast probes investigated in cell labeling applications.4Quantum dots possess good physical and photo stability and have been utilized to facilitate optical tracking of implanted cells.5 As each cell contains many nanoparticles that will be passed down to daughter cells, long-term monitoring of the cell's fate is feasible.
In typical indirect cell labeling, cells to be implanted are transfected with reporter genes and can either generate traceable activities or produce accumulations of traceable products. Traditionally, this has been achieved by using reporter genes that generate fluorescent proteins,6 such as bioluminescent enzymes, or proteins with radioisotope-retaining mechanisms, such as herpes simplex virus thymidine kinase, dopamine type 2 receptor and sodium iodide symporter. These reporter genes using small molecule substrates6–8 have been summarized in several excellent review articles and will not be discussed here. Ferritin and MagA are able to transfer endogenous iron or iron supplements into iron oxide nanoparticles and have been evaluated as MRI reporter genes.9 The use of ferritin and MagA reporter genes for cell tracking will be briefly discussed.
In this article, we will discuss direct and indirect cell labeling approaches with a focus on the former. In particular, we will emphasize iron oxide and quantum dot based cell labeling, which are widely used to gain understanding in the migration and in vivo fate of cell implants.
2. The importance of understanding the fate of cellsin vivo
Stem cells and progenitor cells are under intensive investigations for their self-renewal feature and the ability of differentiating into a diverse range of specialized cell types.10 Such characteristics enable the cells to correct or replace defective cell populations, which, if harnessed well, can revolutionize therapeutics. The most promising applications include treating CNS-related diseases, such as spinal cord injury, Parkinson's disease, myelin disorders, and Huntington's disease as well as myocardial regeneration.11 In the battlefield against cancer, immune cell based therapy has attracted great attention for their role in the control of cancer malignancy. It is expected that by exploiting and augmenting the antitumor power of immune cells, such as T cells, natural killer (NK) cells, B cells and dendritic cells, one could arrive at a novel therapeutic avenue that is more selective than the conventional methods.The ability to monitor cell survival, migration and differentiation is essential for the success of cell based therapies. Certain sized nanoparticles allow efficient particle-cell interaction. The unique optical and/or magnetic properties of the nanoparticles permit non-invasive, accurate and real-time cell tracking.12 In particular, the prolonged cellular retention time of these nanomaterials provides a large observation window and therefore makes it possible for longitudinal cell tracking in vivo.
3. Cell labeling strategies: pros and cons of direct labeling and indirect labeling
In general, there are two approaches for cellular labeling and tracking: direct and indirect cell labeling. Both direct and indirect cell labeling methods are useful in cell therapeutics,13 and each method has its distinct advantages and limitations. Direct labeling is comparatively easy, less expensive and a well-established methodology where the cells are labeled with probes and detected by relevant imaging modalities. Indirect labeling by means of a reporter gene transfection allows for steady, stable, and safe visualization of cellular trafficking, persistence, proliferation, and function at the target site.14 In a direct labeling protocol, high-contrast cell-to-medium or -tissue ratios are achieved easily. However, this approach is limited by a relatively short observation period, since the signal diminishes as the cells proliferate and the marker decays with mitotic division. On the other hand, indirect labeling of cells allows sustained cellular trafficking but demands genetic engineering, which needs sophisticated modification and may be difficult for clinical translation.4. Reporter gene based cell trafficking
Reporter gene (RG) based cellular labeling and imaging involves an indirect coupling of a reporter gene with a complimentary reporter probe.15,16 Delivery of a RG to the target tissue by transfection or transduction is a key requirement for all RG based cellular imaging studies.17–19 A typical RG construct involves a transcriptional control component which functions as a biomolecular genetic sensor. This sensor can respond to endogenous transcription factors and transcription regulating complexes that instigate and regulate RG expression.20,21Ferritin is the primary intracellulariron storage protein which keeps the iron in a soluble and non-toxic form.22 Ferritin plays a critical role in iron homeostasis by chelation and neutralization of labile iron. The iron nanoparticles, stored in the ferritin cages, are either in an antiferromagnetic or superparamagnetic23 form which results in an unique effect on the relaxation of water as detected by MRI. MRI has been used to detect: 1) changes of ferritin content in the heart, liver, spleen and brain, 2) gender-related accumulation of brain iron, and 3) basal ganglia iron accumulation in Parkinson's, Alzheimer's, and Huntington's diseases24
Ferritin has also been studied as a MRI reporter gene. Cells transfected to overexpress H-ferritin (heavy chain ferritin), L-ferritin (light chain ferritin), or combination of H-ferritin and transferrin receptor have been reported.25 In a study by Liu et al.,26 mouse embryonic stem (mES) cell lines carrying human ferritin heavy chain (hFhc) as a RG were used to monitor the cell grafts in vivo using MRI. HFhc generates MRI contrast through compensatory upregulation of transferrin receptor and subsequently increased cellular iron storage in ferritin bound form. Expression of hFhc does not interfere with stem cell pluripotency as observed by effective neural differentiation and teratoma formation. When grafted into CD1 nude mice, hFhc transfected stem cells showed a higher transverse relaxation rate than the wild-type control (Fig. 1). Recently Wang, et al.27 reported a study where ferritin was used in combination with iron oxide nanoparticles in an attempt to achieve a longer cell trafficking period. They proposed that the overexpressed ferritin proteins could sequester iron atoms degraded from iron oxide, so as to improve the duration of cell tracking. To test the concept, a plasmid of murine ferritin H-chain was constructed and transfected into C6 rat glioma cells. Ferritin H-chain gene-transfected C6 and parental C6 cells were each labeled with iron oxide nanoparticles and inoculated bilaterally into nude mice. T2-weighted MRI was performed at selective time points after cell implantation, and the iron content in xenografts was measured. They found that the ferritin overexpressed C6 cells accumulated more iron than parent C6 cells, even though both were labeled with iron oxide. The cooperative effect led to reduced signal intensity in T2-weighted images and extended the monitoring window.
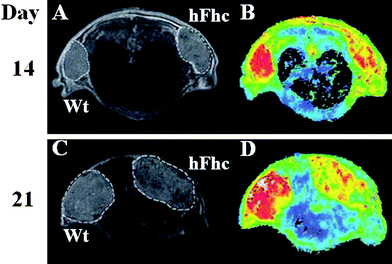 | ||
| Fig. 1 In vivo MRI detection of human ferritin heavy chain (hFhc) transgene induced MRI contrast in mES cellgrafts. (A, C) Representative T2 weighted fast spin echo images showing the same pair of tumors grown from mES grafts (left, wild type(Wt)); right, hFhc transgenic), at days 14 and 21 post inoculation. (B,D) Corresponding color coded T2 maps from multi echo measurements showed significant reduction of T2 relaxation time in the tumor overexpressing hFhc transgene at both time points. (Reprinted with permission from ref. 26). | ||
MagA, a protein found in the magnetotactic bacterium has been studied as a potential candidate for reporter gene imaging.28 MagA is involved in the regulation of iron transport in magnetotactic bacterium, which has the function of transferring Fe ions into magnetite (Fe3O4) nanocrystals. The expression of MagA leads to natural synthesis of nanoparticle-containing magnetosomes that are 20–60% more effective in inducing T2* contrast than ferritin. Zurkiya et al.29 showed that MagA itself was sufficient in producing magnetic nanoparticles in mammalian 293FT cells, and the increased transverse relaxivity can be measured by MRI. Goldhawk et al.30 expressed MagA in mouse neuroblastoma N2A cells which created intracellular contrast detectable by 11T MRI. Results also showed that MagA activity in mammalian cells responded to iron supplementation and functioned as a contrast agent that could be deactivated by a single point mutation.
One limitation of the reporter gene based imaging method is that the strong overexpression of transgenes may cause cytosolic, nuclear and membrane accumulation of reporter gene products.31 Also cell surface transporters and receptors need the incorporation of these proteins into the plasma membrane in order to institute and preserve their reporter function. This could potentially lead to a change in the biology of transduced cells by enhanced contact to endogenous substrates and ligands mediated through the expression of the transduced receptor, transporter or enzyme. There are also concerns about the potential signaling and the cell biology effects of using receptors or transporters as reporter genes. It is likely that human reporter gene imaging will utilize more than one reporter system to monitor the cell fate.32–34
5. Nanoparticles for cell labeling
Nanoparticles 35,36 with large surface area to volume ratio facilitate surface functionality.37–40 The unique physicochemical properties of nanoparticles allow visualization of cellsin vivo using different molecular imaging modalities.41,42 such as magnetic resonance imaging (MRI), radionuclide imaging (positron emission tomography (PET) and single-photon emission computed tomography (SPECT)) and optical imaging. Each of these imaging modalities differs in their sensitivity and resolution.43 The nanoparticles can be either passively introduced (non-targeted) or actively introduced (targeted, see Table 1)44–47 into cells of interest. Nanoparticle probes48,49 used in each of these imaging modalities will be discussed in the following sections.| Biomarker | References |
|---|---|
| a EGF- Epidermal growth factor; VEGF- Vascular endothelial growth factor, TGF- Transforming growth factor; IGF- Insulun like growth factor. | |
| Integrins (α5β3, α5β5) | 44,45,46 |
| Growth factors (EGF, VEGF, TGF, IGF) | 45,47 |
| Endosialin | 44 |
| Folate receptor | 45,46 |
| Fibronectin | 44 |
| Interleukins | 44 |
| Transferrin | 45,46 |
| Nucleolin | 44 |
| Aminopeptidase N | 44 |
| Annexin A1 | 44 |
| Collagen | 44 |
| Prostate specific membrane antigen | 45 |
| Galactosamine | 45 |
5.1 MR labeling
Anatomical and functional MRI50–53 is a powerful imaging modality offering excellent soft tissue contrast, imaging in oblique orientations, and collecting data in two and three dimensions.54,55 MRI is a non-invasive diagnostic deep tissue imaging modality that generates cross-sectional images of the human body. MR imaging makes use of a powerful magnetic field to align the nuclear magnetization of hydrogen atoms of water in the body. Radio frequency with its signal homogeneity and sensitivity systematically alters the alignment of the magnetization, causing the hydrogen nuclei to produce a rotating magnetic field detectable by the scanner. The signal obtained can then be manipulated by additional magnetic fields to construct a whole body image.56MR imaging to track cells combines the ability of MRI with contrast probes for labeling cells,57–59 providing dynamic measurement of cell migration into target tissues. MR contrast probes in nanomolar to micromolar concentrations can alter the relaxation rates of many nearby tissue water protons making them conspicuous on post contrast enhanced MRI. Changes in relaxivity will affect either the T1 (spin–lattice) or T2 (spin–spin) relaxation. Paramagnetic probes mainly affect T1 while the superparamagnetic probes are known to affect T2 relaxation.60–64
The most straightforward strategy to improve cell uptake rate is to increase the initial particle concentration. This works to some extent with phagocytic cells, such as T cells,65,66monocytes,67 and macrophage68,69cells, but not for non-phagocytic cell types. To address that, a variety of uptake-facilitating agents have been introduced to the nanoparticle surface. For instance, HIV tat peptide has been covalently bound to the particle surface to increase the transportation across the cell membrane.70,71 The resulted nanoparticles were able to induce 10–30 pg Fe/cell uptake with hematopoietic and neural progenitor cells.71 Also internalizing monoclonal antibodies (mAbs), such as mouse anti-transferrin mAb OX-26 and anti-CD11 mAb, have been conjugated onto iron IONPs to promote labeling of oligodendrocyte and dendritic cells.72,73 However, these methods require bioconjugation techniques and are economically unfavorable. Especially in the case of receptor mediated internalization, only cells expressing the particular receptor targets can be effectively labeled, which limits general use of the particle conjugates.
A more convenient route to enhance uptake is to co-apply with the particle excipients. Examples include poly-L-lysine (PLL), protamine syulfate, dendrimers, lipofectamine and PEI, which are highly positively charged, and can complex with IONPs to promote their shuttling across the anionic cell membrane. In this approach, the complex formation is achieved in situ through simple mixing and agitation. As compared to receptor mediated internalization, this approach is not species-specific, and hence can be applied to a wide range of cell lines. Taking PLL-Feridex labeling as an example, an iron uptake of 10–20 pg per cell can be achieved with many cell lines.74 However, these excipients are usually toxic at high concentrations and the tolerance may vary among different cell types. Therefore, optimization of transfection and nanoparticle combination has to be made with each cell line to achieve highest uptake rate while not affecting the cell growth and function. Also, synthetic approaches have been developed to synthesize IONPs, where polymers are introduced into the formula during the synthesis rather than after the particle formation. PLL and dendrimers, for instance, have been used as capping ligands to yield IONPs conjugates that can be directly applied in cell labeling. In particular, a generation 4.5 carboxylated dendrimer was used to generate iron oxide nanoparticles, and the resulted conjugates have been applied in labeling a number of cell lines with similar labeling rate.63
Particles made from pyrolysis methods have also been used for cell labeling, with the premise, that they are surface modified and water soluble. One common strategy to achieve such phase conversion is to add one layer of amphiphilic materials, which interact with the existing alkyl coating and suspend the particles with their out-exposed hydrophilic section. For instance, alkylated PEI was utilized as a surface modification material, which can anchor onto hydrophobic iron oxide nanoparticle surface via hydrophobic-hydrophobic interactions. The nanoconjugates coated with one layer of polycation PEI can be efficiently internalized by various cell lines. More interestingly, when lowering the starting ratio of alkylated PEI and IONPs, several IONPs can be encapsulated into a single micelle-like structure, and the resulting nanoclusters showed much improved R2 relaxivity and labeling efficiency.75 Aside from such a surface-addition approach, the hydrophobic IONPs can be converted water soluble by ligand exchange. Ligands with high affinity to the particle surface are applied, which replace the original coating layer making the particle surface hydrophilic. Xie et al.76 developed a two-step surface treatment to modify the particle surface. In the first step, dopamine, with its two adjacent hydroxyl groups, chelated with the surface iron and replaced the alkyl coating. The resulting dopamine coated IONPs were not readily water soluble but could be well dispersed in polar organic solvents such as DMSO. In the second step, nanoparticles in DMSO were added dropwise to human serum albumin (HSA) in aqueous solution, a recipe that resembles the formation of Abraxane. With an excellent ligand binding characteristic, the HSA molecules were adsorbed onto the nanoparticles providing extra stability against aggregation. Such nanoparticles are efficient in labeling a variety of cell lines presumably due to the incompletely shielded, amine-rich dopamine layer. Unlike many other iron oxide based cell labeling formulas that rely on polycation polymers to induce efficient endocytosis, such dopamine-HSA coated nanoparticles comprise only aminated small molecules and have much less concern in toxicity (Fig. 2).
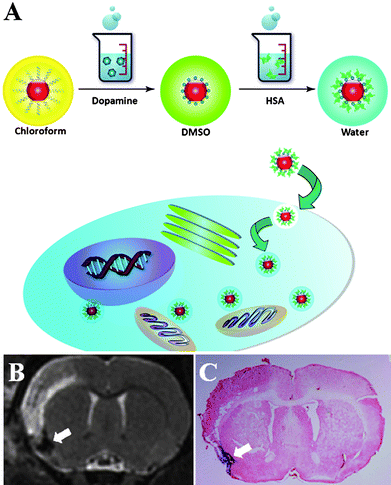 | ||
| Fig. 2 (A) Cartoon illustrating the human serum albumin coated iron oxide nanoparticle formation and cell labeling. (B) Longitudinal MRI studies (day 7) and (C) Prussian blue staining on a rat focal cerebral ischemia model injected with iron-laden macrophages. (Reprinted with permission from ref. 76). | ||
The Fe loading rate for most IONP formulas falls between 10 to 20 pg/cell. Some unusual treatments may be needed to achieve higher labeling efficiency. For example, magnetofection takes advantage of the magnetic nature of iron oxide nanoparticles. It builds a magnetic field upon the to-be-transfected cells, which is believed to induce increased cell membrane permeability and improve cell translocation. Also, micro-sized particles were found to be advantageous in cell labeling.77 The increase in size may have a negative effect on the particle's internalization rate, but since each single particle contains sufficient amount of iron, the final iron loading efficiency can be as high as 100 pg Fe/cell. It was reported that at such iron level it is possible to detect a single cell with MRI.78
In another study, Ghaghad et al.80 reported a liposomal-based Gd nanoparticle formula as a MR contrast agent, named dual-Gd liposomal agent. This novel nanoparticle features two agents, CE-Gd (core-encapsulated Gd liposome) and SC-Gd (surface-conjugated Gd liposome) to create an entity that is capable of delivering a higher concentration of Gd thus greater signal enhancement per particle compared to core-encapsulated and surface-conjugated Gd liposomal agents separately. The dual-Gd liposomal agent was tested in vivo for contrast-enhanced magnetic resonance angiography (CE-MRA) studies. Dual-Gd liposomes demonstrated higher T1 relaxivity and higher signal-to-noise and contrast-to-noise ratios in CE-MRA studies than the core-encapsulated or the surface-conjugated liposomal agents alone. This dual-Gd liposomal MRI agent could find applications in molecular imaging.
Manganese is another well-studied T1 contrast agent. Manganese enhanced MRI has been used to study neuronal activity, monitor neuronal tracts, and neuronal connectivity in animal models of disease, as well as to monitor liposomal drug delivery.81 In a recent study by Gilad et al.,82MnO nanoparticles were evaluated as a novel T1 contrast agent for cell labeling. In this report MnO nanoparticles were successfully used to detect cells with positive contrast in vivo and multiplexed with iron oxide to detect two cell populations simultaneously with opposite cell contrast.
5.2. Optical labeling
Optical labeling (OL) is based on the detection of fluorescence. The method is based on ex vivolabeling of cells with a fluorescent tag, subsequent engraftment of the labeled cells and visualization of their accumulation in specific target organelles of interest. OL is as sensitive as radiolabel based imaging techniques, but without any exposure to irradiation.83 OL provides an effective means of non-invasively tracking cells repeatedly, thereby providing insight into cell migration to the target site.6 The cell labeling efficiency is usually improved if the cells are incubated with the fluorescent dye in serum free media as opposed to incubation in serum containing media. One major downside of OL is the limited tissue penetration of fluorescent labels in vivo with a tracer accumulation in deep tissues, more than about 4–10 cm from the skin surface, may not be detected.84 Availability of tailor made fluorescent probes has made fluorescence imaging of optically labeled cells as one of the most utilized optical imaging modalities.85,86 Nanomaterial based cellular labels like quantum dots have made OL a relatively low-cost method and has become an indispensable tool in small animal studies.87One of the major applications of fluorescent probes in the biological field has been labeling cell structures.92 QDs are preferred over organic fluorophores for multicolor cell labeling because of their high quantum yields, high molar excitation coefficients, strong resistance to photobleaching and chemical degradation, broad excitation spectra, long fluorescence lifetimes, narrow emission spectra and large stokes shift.93–101 Alivisatos and Nie research groups were the first to demonstrate QDs as luminescent cell markers.102,103 QDs are ideal candidates for multiphoton microscopy in live animals since the product of the nonlinear two-photon absorption cross section and the fluorescence quantum yield provide a direct brightness measure for imaging. Modeling studies have also revealed that two spectral windows, 700–900 nm and 1200–1600 nm, exist for optimal QD-based optical imaging in vivo.104–116
In a study by Slotkin et al.,117 direct QD labeling of mammalian stem and progenitor cellsin vivo was carried out. In this work in uteroelectroporation and ultrasound guided in vivo delivery techniques were developed to specifically and efficiently label neural stem and progenitor cells (NSPCs) of the developing mammalian central nervous system with QDs. Direct labeling of NSPCs was achieved using ultrasound guided biomicroscopy (UBM) injection of 620 nm COOH-conjugated QDs into the parenchyma of the ventral telencephalon of mouse embryos. QDs were found in principle cell types generated by NSPCs at substantial distances away from the initial site of injection, suggesting that QD loading of NSPCs in vivo did not inhibit their migration or differentiation during the developmental cycle. Using in utero electroporation, NSPCs in viable mouse embryos were labeled with QDs. The ventricular neuroepithelium of mouse embryos was coelectroporated with a mix of 620 nm COOH-functionalized QDs and EGFP-F plasmid. This enabled a 3D reconstruction of QD loaded ventricular and subventricular zones of neural precursor cellsin situ using multiphoton imaging. These studies established a novel approach to study the developing mammalian embryo and CNS. As seen by these techniques, QDs can be effectively used to label mammalian NSPCs in vivo and study various biological processes involved during mammalian development.
Dendritic cells (DCs) play a key role in initiating an adaptive immune response by presenting antigen to T cells in lymphoid organs. In a recent study by Noh et al.,118 DCs were labeled with QDs for imaging their migration into lymph nodes in vivo. In brief, QD-labeled DCs were injected into the footpad, and their migration into popliteal and inguinal lymph nodes was observed via NIR fluorescence imaging (Fig. 3). Labeling of DCs with QDs showed no effect on DC phenotype or maturation potential, as was observed by MTT assay and FACS analysis. Lim et al.119 labeled natural killer (NK) cells with QDs and monitored the cell based therapeutics with fluorescence imaging. In this study, QD705 were coupled with anti-human CD56 antibody, and the conjugates were utilized to label human natural killer cells (NK92MI). FACS analysis showed that the QD labeling had no effect on the NK92MI cell viability. Anti-human CD56 antibody-coated QD705 labeling on the NK92MI cell function was investigated by measuring interferon gamma (IFN-γ) production and cytolytic activity. The QD labeled NK92MI cells showed a therapeutic effect similar to that of unlabeled NK92MI cells. These results demonstrate that QD based cell labeling can be used as a safe and effective means to facilitate in vivo trafficking of therapeutic cells.
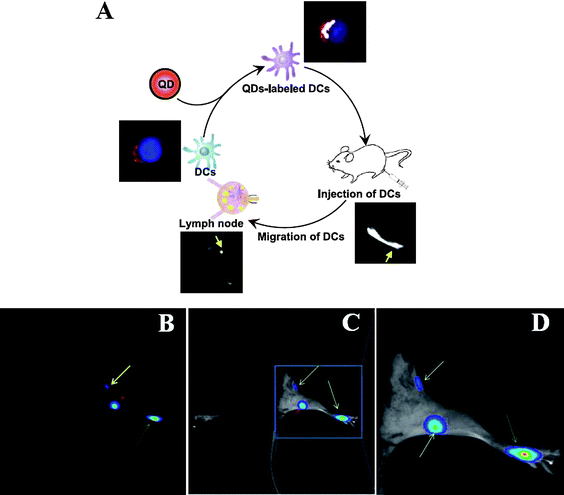 | ||
| Fig. 3 (A) Schematic showing overview after dendritic cells (DCs) were cocultured and labeled with NIR-emitting QDs. QD-labeled DCs 1 × 106cells/50 μl were injected into the hind-leg footpad of the mouse, and the migrations of injected DCs into lymph nodse were monitored by in vivo NIR imaging system. After 2 days, the mouse was anesthetized, depilated and imaged. (B-C) Strong NIR fluorescence signals (pseudocolor) are clearly observed in the right foot (injection site; green arrow), popliteal lymph node (red arrow) and inguinal lymph node (yellow arrow) of the mouse injected with QD-labeled DCs. (D) Enlarged image of lymph node part in C. (Reprinted with permission from ref. 118). | ||
5.3. Multimodality imaging
Ideally, cell imaging and labeling can gather information at both anatomic and functional levels with high spatial and temporal resolution.126–128 Unfortunately, no single modality is sufficient to provide all these details as each modality has its own limitations. For instance, MRI has high resolution yet it suffers from low sensitivity. Radionuclide-based imaging techniques have very high sensitivity but have relatively poor spatial resolution. It is difficult to acquire quantitative fluorescence analysis with living subjects, particularly for deep tissues.129 The combination of several molecular imaging modalities can offer synergistic advantages over any one modality alone. Combining optical imaging modality with 3D tomographic techniques such as PET, SPECT, or MRI can allow for noninvasive imaging in living subjects with higher sensitivity and/or accuracy with the needed resolution.130–132 Multimodality imaging has emerged133 as a technology that utilizes the strengths of different modalities and yields a hybrid134,135 imaging platform.136 Hence multimodality cell imaging integrates two or more cellular imaging modalities.137 NPs having large surface areas allow multiple functional moieties to be incorporated for multimodality molecular imaging.138,139Multimodality imaging probes enable multiple in vivo imaging molecular readouts simultaneously detected (via optical, MRI and/or nuclear modalities, for instance), and enable in vivo imaging and concomitant targeted therapy.140,141 In a recent study, Shi et al.142 developed bifunctional anionic Eu3+-doped Gd2O3 hybrid nanoparticles as a luminescent and T1 weighted MRI contrast agent for stem cell labeling. The Gd2O3:Eu nanoparticles were synthesized through the polyol method and further modified with citric acid to obtain anionic nanoparticles. Cellular uptake of these nanoparticles into hMSCs was confirmed by confocal laser scanning microscopy after two hours of nanoparticle incubation. Image comparison of labeled cells and unlabeled control clearly showed the presence of enhanced fluorescent signals uniformly distributed in the cytoplasm. The amount of Gd internalized in the hMSCs was quantified by ICP-MS. The labeling process had no adverse effect on the viability, proliferation, osteogenic, adipogenic and chondrogenic differentiation potential of the hMSCs. The reported data indicate that the biocompatible anionic Gd2O3 nanoparticles doped with Eu3+ show promise both as a luminescent and T1 contrast agent for use in visualizing hMSCs. Vuu et al.143 reported a rhodamine (fluorescent) and gadolinium (paramagnetic) dually labeled polymerized nanoparticle, which was found to efficiently label tumor cells. The nano constructures were prepared from lipid monomers with diacetylene bonds that were sonicated and photolyzed to form polymerized hybrid nanoparticles. Such Gd-Rd-NPs offer in vivo T1-weighted cellular imaging by MRI and detailed histological analysis through fluorescence microscopy. Cells labeled with Gd-Rd-NPs were viable and demonstrated little or no sign of altered cellular morphology. Labeled tumor cells were inoculated subcutaneously into the flanks of C3H mice and imaged via MRI and optical fluorescence imaging modalities (Fig. 4). Results suggest that these nanoparticles could be used to track cellsin vivo. The authors concluded that this basic platform could be modified with different fluorophores and targeting agents for the trafficking of metastatic cells, stem cells and immune cells.
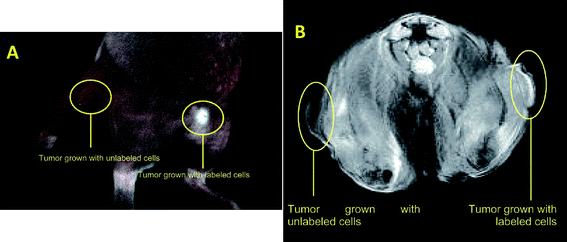 | ||
| Fig. 4 A. In vivo optical imaging (Maestro In Vivo Multispectral Imaging System; Woburn, MA) of mouse hind limb after inoculation with unlabeled tumor cells or gadolinium-rhodamine nanoparticle-labeled tumor cells. A female C3H mouse bearing unlabeled tumor (left flank) and Gd-Rd-NP-labeled tumor (right flank) imaged 7 days post subcutaneous inoculation. Note that the fluorescent signal is localized only to the labeled tumor site and that no signal was received from the unlabeled (control) tumor. B. Axial MRI section of mouse 7 days after inoculation with unlabeled tumor cells and gadolinium-rhodamine nanoparticle-labeled tumor cells. Marked contrast enhancement can be visualized in the Gd-Rd-NP-labeled tumor vs. unlabeled (control) tumor. (Reprinted with permission from ref. 143). | ||
Patel et al.144 developed an ion-sensing nanoparticle formula comprised of a superparamagnetic iron oxide (SPIO) core encapsulated within a porous silica shell. The latter could be readily anchored with ligands capable of coordinating with positron-emitting metals. This hybrid nanoparticle has great potential for use in cell tracking with both MRI and PET functions. Rat adult stem cells were used to study the cell-labeling efficacy, cytoxicity and relaxivity in comparison to one of the most commonly utilized MRI contrast agents, Feridex. The IO-silica hybrid nanoparticle probe with clustered iron oxide cores displayed relaxivities comparable to Feridex. The nanoparticles also demonstrated excellent biocompatibility, exhibiting no short-term cytotoxicity to the stem cells with high cell uptake efficiency. In another study reported by Luet al.,145 bifunctional superparamagnetic SPIO@SiO2(FITC) core/shell nanoparticles were synthesized for dual modal detection of bone marrow hMSCs. This study demonstrated that the SPIO@SiO2(FITC) nanoparticles could be efficiently internalized into hMSCs. The procedure used to label hMSCs took 30 min to 1 h incubation time and was easy to apply. The concentration of SPIO@SiO2(FITC) nanoparticles for labeling cells was low and the labeled cells could be visualized in a clinical 1.5T MR imager with a detection limit of 1.2 × 105cellsin vivo and 104cellsin vitro (Fig. 5). Park et al.146 reported silica-coated magnetic nanoparticles incorporating rhodamine B isothiocyanate, MNPs@SiO2(RITC) for multimodal visualization of hMSCs in vivo. In this study hMSCs were labeled with MNPs@SiO2(RITC) and subcutaneously injected into NOD.CB17-PrkdcSCID (NOD-SCID) mice. The transplanted hMSCs were successfully detected using MR and optical imaging suggesting MNPs@SiO2(RITC) can be applied for multimodal cell labeling and tracking. Multimodal MRI and bioluminescent evaluation of stem cell viability delivered into different zones of myocardial infarction was studied by Chung et al.147 In this study mESCs were dual labeled with superparamagnetic IONPs and luciferasevia direct injection into 3 different zones of myocardial infarction. MRI was used to assess the precise anatomic localization of dual-labeled mESCs. Bioluminescent imaging enabled assessment of the viability of mESCs transplanted into the infarct myocardium. This data showed that multimodal evaluation of dual labeled mESC engraftment in the heterogeneous tissue of infarcted myocardium is possible. In a similar study, Higuchi et al.148 reported the localization and viability assessment of transplanted cells using genetic labeling with the human sodium iodine symporter gene and magnetic labeling with IONPs. Multimodality imaging of human endothelial progenitor cells with PET and MRI using magnetic and genetic dual cell labeling allowed for the simultaneous assessment of cell localization and viability after transplantation.
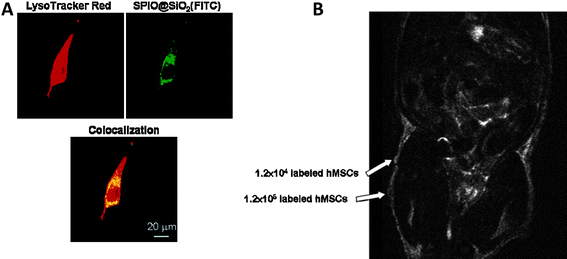 | ||
| Fig. 5 A. Colocalization of green fluorescent SPIO@SiO2(FITC) with late endosomes/lysosomes. hMSCs were treated with 30 μg/mL SPIO@SiO2(FITC) for 30 min and then incubated with LyosTracker Red for another 30 min. The cellular distribution of SPIO@SiO2(FITC) and LysoTracker Red-labeled organelles (late endosomes/lysosomes) were analyzed by a Zeiss Axiovert 100M confocal unit. B. T2 weighted MRI study of SPIO@SiO2(FITC)-labeled hMSCs was performed by injecting 1.2 × 104 or 1.2 × 105cells mixed with Matrigel into subcutaneous tissue of the flanks (unlabeled cells at left flank and labeled cells at right flank) of a nude mouse. (Reprinted with permission from ref. 145). | ||
6. Nanoparticle toxicity
Toxicity is the least understood feature of nanoparticles when it comes to its application for in vivo imaging. Once introduced in to the system toxicity can occur from the NPs themselves or from the associated components of the NPs that might get released during degradationin vivo. Traditionally QDs are composed of cadmium salts which can release soluble cadmium after cellular labeling leading to toxicity though some reports suggests surface modification of the QDs can address this issue.149 Absorption, distribution, metabolic clearance, and excretion characteristics are highly variable for QDs because of the wide variation in QD physicochemical properties making QDs unlikely to be approved for in vivo imaging in humans unless substantial safety evaluation is shown. One approach could be to include more benign elements instead of Cd in the formulation like CuInS2/ZnS core/shell QDs which do not contain any Class A elements or Class B elements which may show great potential as biocompatible probes for in vivocell labeling.150 Toxicity of the NPs can be due to several other factors such as concentration, hydrodynamic size, surface charge and type of coatings and the route of administration. IONPs come in different sizes ranging from 10–300 nm with various surface coatings.151 In a recent study PEI coated IONP showed a dose dependent toxicity on cell viability of baby hamster kidney BHK21 cellsin vitro.152In vivoIONP toxicity has resulted from the surface charge of the coated material along with non-specific cellular and tissue biodistribution.153,154 Aggregation of the IONPs in presence of a magnetic field in vivo could lead to problems like embolisation.155 Another major limitation of the IONP is their uptake by phagocytic cells leading to rapid clearance from the blood and excessive IONP could also lead to an imbalance in its homeostasis and may lead to toxicity.156 Some of the issues could be addressed by choosing the appropriate size and coating materials. Most popular surface coating so far has been dextran for IONPs. Recently PEG, chitosan and polyvinyl alcohol has also been incorporated in the formulation which has shown to address some of the above mentioned issues involving IONPs.157,158 Similarly carbon, gold, silver, manganese, gadolinium based nanomaterials have shown toxicity owing to agglomeration, surface chemistry, dosage and way of administration.159 Again the emphasis is on selecting the right size and biocompatible coating being the major factors, though long term exposure and biodistribution studies in vivo are a must before implementing any of the nanoformulations in to practice.7. Conclusions and perspectives
Nanoparticle-based cell labeling has emerged as an important aspect of current disease diagnostics and therapeutics. Tracking and monitoring transplanted cells using nanoparticles has shown promising results in vitro and in vivo. Because of their unique mechanical, optical and electrical properties, nanoparticles could be used for efficient labeling of stem, immune, and cancer cells. Ferritin nanoparticle based cell labeling is a potential candidate as a reporter gene for detection of transcriptional regulation of gene expression by MRI. Superparamagnetic iron oxide nanoparticles have emerged as the most widely adopted cell labeling probes for T2 based MR imaging. Gadolinium and manganese based nanoparticle formulations are increasingly used as T1 based MRI contrast agents. As far as optical modality is concerned, QDs remain the most sought out cell labeling probe so far. QDs with tailor made surfaces have been utilized to track stem cells as well as cancer cells. Other fluorescent optical probes such as silica, gold and other nanomaterials are also being tested as efficient cell labeling probes. With the infusion of various imaging modalities it is now possible to collect a variety of data from a single set of experiments.Even though various nanoparticle probes are now available for cellular labeling for efficient tracking in vivo, some toxicity issues affiliated with the nanoparticle probes such as metal content and aggregation behavior need to be addressed in detail before they can be translated into clinic.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, including the National Institute of Biomedical Imaging and Bioengineering.References
- M. K. Carpenter, J. Frey-Vasconcells and M. S. Rao, Nat. Biotechnol., 2009, 27, 606–613 CrossRef CAS.
- J. Marin-Garcia and M. J. Goldenthal, Curr. Stem Cell Res. Ther., 2006, 1, 1–11 Search PubMed.
- L. S. Hart and W. S. El-Deiry, J. Clin. Oncol., 2008, 26, 2901–2910 CrossRef.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- F. Kiessling, Handb. Exp. Pharmacol., 2008, 305–321 Search PubMed.
- V. Ponomarev, J. Nucl. Med., 2009, 50, 1013–1016 CrossRef CAS.
- M. Modo, Curr. Opin. Organ Transplant, 2008, 13, 654–658 Search PubMed.
- A. A. Gilad, K. Ziv, M. T. McMahon, P. C. M. van Zijl, M. Neeman and J. W. M. Bulte, J. Nucl. Med., 2008, 49, 1905–1908 CrossRef CAS.
- K. R. Boheler, J. Czyz, D. Tweedie, H.-T. Yang, S. V. Anisimov and A. M. Wobus, Circ. Res., 2002, 91, 189–201 CrossRef CAS.
- C. Corot, P. Robert, J. M. Idee and M. Port, Adv. Drug Delivery Rev., 2006, 58, 1471–1504 CrossRef CAS.
- J. V. Frangioni, J. Clin. Oncol., 2008, 26, 4012–4021 CrossRef.
- V. Ponomarev, J. Nucl. Med., 2009, 50, 1013–1016 CrossRef CAS.
- S. Gambhir, H. Herschman, S. Cherry, J. Barrio, N. Satyamurthy, T. Toyokuni, M. Phelps, S. Larson, J. Balaton, R. Finn, M. Sadelain, J. Tjuvajev and R. Blasberg, Neoplasia, 2000, 2, 118–138 CrossRef CAS.
- I. Y. Chen, J. M. Greve, O. Gheysens, J. K. Willmann, M. Rodriguez-Porcel, P. Chu, A. Y. Sheikh, A. Z. Faranesh, R. Paulmurugan, P. C. Yang, J. C. Wu and S. S. Gambhir, Mol. Imaging Biol., 2009, 11, 178–187 Search PubMed.
- C. Martelli, I. V. Libani, R. Lui and L. Ottobrini, Minerva Biotecnologica, 2009, 21, 53–66 Search PubMed.
- A. De, X. Z. Lewis and S. S. Gambhir, Mol. Ther., 2003, 7, 681–691 CrossRef CAS.
- R. H. Unger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12641–12642 CrossRef CAS.
- J. H. Kang and J.-K. Chung, J. Nucl. Med., 2008, 49, 164S–179 CrossRef CAS.
- R. G. Blasberg, Neuropsychopharmacology, 2004, 29, S25–S25.
- H. R. Herschman, in Advances in Cancer Research, 2004, vol. 92, pp. 29–80 Search PubMed.
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- J. M. Domínguez-Vera, B. Fernández and N. Gálvez, Future Med. Chem., 2010, 2, 609–618 Search PubMed.
- G. Bartzokis, T. A. Tishler, P. H. Lu, P. Villablanca, L. L. Altshuler, M. Carter, D. Huang, N. Edwards and J. Mintz, Neurobiol. Aging, 2007, 28, 414–423 CrossRef CAS.
- B. Cohen, K. Ziv, V. Plaks, A. Harmelin and M. Neeman, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2009, 1, 181–188 Search PubMed.
- J. Liu, E. C. H. Cheng, R. C. Long, S.-H. Yang, L. Wang, P.-H. Cheng, J. Yang, D. Wu, H. Mao and A. W. S. Chan, Tissue Eng., Part C, 2009, 15, 739–747 Search PubMed.
- J. Wang, J. Xie, X. Zhou, Z. Cheng, N. Gu, G. Teng, Q. Hu, F. Zhu, S. Chang, F. Zhang, G. Lu and X. Chen, Mol. Imaging Biol., 2010 May 4 Search PubMed.
- J. Xie, K. Chen and X. Chen, Nano Res., 2009, 2, 261–278 CrossRef CAS.
- O. Zurkiya, A. W. S. Chan and X. Hu, Magn. Reson. Med., 2008, 59, 1225–1231 CrossRef CAS.
- D. E. Goldhawk, C. Lemaire, C. R. McCreary, R. McGirr, S. Dhanvantari, R. T. Thompson, R. Figueredo, J. Koropatnick, P. Foster and F. S. Prato, Mol. Imaging, 2009, 8, 129–139 CAS.
- A. A. Gilad, P.T.W. Jr., P. C. M.v. Zijl and J. W. M. Bulte, NMR Biomed., 2007, 20, 275–290 CrossRef CAS.
- A. A. Gilad, P. Walczak, M. T. McMahon, H. Bin Na, J. H. Lee, K. An, T. Hyeon, P. C. M. van Zijl and J. W. M. Bulte, Magn. Reson. Med., 2008, 60, 1–7 CrossRef CAS.
- A. A. Gilad, K. Ziv, M. T. McMahon, P. C. M. van Zijl, M. Neeman and J. W. M. Bulte, J. Nucl. Med., 2008, 49, 1905–1908 CrossRef CAS.
- A. A. Gilad, P. T. Winnard, P. C. M. van Zijl and J. W. M. Bulte, NMR Biomed., 2007, 20, 275–290 CrossRef CAS.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- W. J. Rieter, J. S. Kim, K. M. L. Taylor, H. Y. An, W. L. Lin, T. Tarrant and W. B. Lin, Angew. Chem., Int. Ed., 2007, 46, 3680–3682 CrossRef CAS.
- C. J. Xu, Z. L. Yuan, N. Kohler, J. M. Kim, M. A. Chung and S. H. Sun, J. Am. Chem. Soc., 2009, 131, 15346–15351 CrossRef CAS.
- S. H. Choi, H. Bin Na, Y. Il Park, K. An, S. G. Kwon, Y. Jang, M. Park, J. Moon, J. S. Son, I. C. Song, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2008, 130, 15573–15580 CrossRef CAS.
- H. Wang, F. Cao, A. De, Y. Cao, C. Contag, S. S. Gambhir, J. C. Wu and X. Chen, Stem Cells, 2009, 27, 1548–1558 Search PubMed.
- J. S. Choi, Y. W. Jun, S. I. Yeon, H. C. Kim, J. S. Shin and J. Cheon, J. Am. Chem. Soc., 2006, 128, 15982–15983 CrossRef CAS.
- C. Xu, J. Xie, D. Ho, C. Wang, N. Kohler, E. G. Walsh, J. R. Morgan, Y. E. Chin and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 173–176 CrossRef CAS.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS.
- R. F. Minchin and D. J. Martin, Endocrinology, 2010, 151, 474–481 CrossRef CAS.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS.
- E. Gullotti and Y. Yeo, Mol. Pharmacol., 2009, 6, 1041–1051 CrossRef CAS.
- T. Islam and L. Josephson, Cancer Biomark., 2009, 5, 99–107 Search PubMed.
- S. Zhang and H. Uludag, Pharmacol. Res., 2009, 26, 1561–1580 CrossRef CAS.
- M. Zhou, H. Fei, Y. Liu and W. F. Li, Progr. in Chem., 2010, 22, 201–209 Search PubMed.
- B. Perlstein, T. Lublin-Tennenbaum, I. Marom and S. Margel, J. Biomed. Mater. Res., Part B, 2010, 92B, 353–360 CAS.
- R. Christopher deCharms, Nat. Rev. Neurosci., 2008, 9, 720–729 CrossRef.
- D. L. Kraitchman, W. D. Gilson and C. H. Lorenz, J. Magn. Reson. Imaging, 2008, 27, 299–310 CrossRef.
- S. Kos, R. Huegli, G. M. Bongartz, A. L. Jacob and D. Bilecen, Eur. Radiol., 2008, 18, 645–657 CrossRef.
- H. Saybasili, A. Z. Faranesh, C. E. Saikus, C. Ozturk, R. J. Lederman and M. A. Guttman, J. Magn. Reson. Imaging, 2010, 31, 1015–1019 CrossRef.
- H. Zaidi, M.-L. Montandon and A. Alavi, Magn. Reson. Imaging Clin. N. Am., 2010, 18, 133–149 CrossRef.
- J. C. Richardson, R. W. Bowtell, K. Mader and C. D. Melia, Adv. Drug Delivery Rev., 2005, 57, 1191–1209 CrossRef CAS.
- H. A. Sharma, Acta Neuropsychiatrica, 2009, 21, 200–201 CrossRef.
- J. S. Guthi, S. G. Yang, G. Huang, S. Z. Li, C. Khemtong, C. W. Kessinger, M. Peyton, J. D. Minna, K. C. Brown and J. M. Gao, Mol. Pharmacol., 2010, 7, 32–40 CrossRef CAS.
- A. M. Reddy, B. K. Kwak, H. J. Shim, C. Ahn, H. S. Lee, Y. J. Suh and E. S. Park, J. Korean Med. Sci., 2010, 25, 211–219 Search PubMed.
- M. R. Bernsen, A. D. Moelker, P. A. Wielopolski, S. T. van Tiel and G. P. Krestin, Eur. Radiol., 2010, 20, 255–274 CrossRef.
- J. F. Lau, S. A. Anderson, E. Adler and J. A. Frank, Nat. Rev. Cardiol., 2010, 7, 97–105 Search PubMed.
- T. Atanasijevic, M. Shusteff, P. Fam and A. Jasanoff, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14707–14712 CrossRef CAS.
- J. W. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS.
- M. Modo, M. Hoehn and J. W. Bulte, Mol. Imaging, 2005, 4, 143–164.
- A. S. Arbab, W. Liu and J. A. Frank, Expert Rev. Med. Devices, 2006, 3, 427–439 Search PubMed.
- T. C. Yeh, W. Zhang, S. T. Ildstad and C. Ho, Magn. Reson. Med., 1993, 30, 617–625 CrossRef CAS.
- J. C. Sipe, M. Filippi, G. Martino, R. Furlan, M. A. Rocca, M. Rovaris, A. Bergami, J. Zyroff, G. Scotti and G. Comi, Magn. Reson. Imaging, 1999, 17, 1521–1523 CrossRef CAS.
- M. L. Zelivyanskaya, J. A. Nelson, L. Poluektova, M. Uberti, M. Mellon, H. E. Gendelman and M. D. Boska, J. Neurosci. Res., 2003, 73, 284–295 CrossRef CAS.
- R. Weissleder, H. C. Cheng, A. Bogdanova and A. Bogdanov Jr, J. Magn. Reson. Imaging, 1997, 7, 258–263 CrossRef CAS.
- A. Moore, R. Weissleder and A. Bogdanov Jr, J. Magn. Reson. Imaging, 1997, 7, 1140–1145 CrossRef CAS.
- L. Josephson, C. H. Tung, A. Moore and R. Weissleder, Bioconjugate Chem., 1999, 10, 186–191 CrossRef CAS.
- M. Lewin, N. Carlesso, C. H. Tung, X. W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS.
- J. W. Bulte, T. Ben-Hur, B. R. Miller, R. Mizrachi-Kol, O. Einstein, E. Reinhartz, H. A. Zywicke, T. Douglas and J. A. Frank, Magn. Reson. Med., 2003, 50, 201–205 CrossRef.
- E. T. Ahrens, M. Feili-Hariri, H. Xu, G. Genove and P. A. Morel, Magn. Reson. Med., 2003, 49, 1006–1013 CrossRef CAS.
- J. A. Frank, B. R. Miller, A. S. Arbab, H. A. Zywicke, E. K. Jordan, B. K. Lewis, L. H. Bryant Jr and J. W. Bulte, Radiology, 2003, 228, 480–487 Search PubMed.
- Z. Wang, G. Liu, J. Sun, B. Wu, Q. Gong, B. Song, H. Ai and Z. Gu, J. Nanosci. Nanotechnol., 2009, 9, 378–385 CrossRef CAS.
- J. Xie, J. Wang, G. Niu, J. Huang, K. Chen, X. Li and X. Chen, Chem. Commun., 2010, 46, 433–435 RSC.
- E. M. Shapiro, S. Skrtic and A. P. Koretsky, Magn. Reson. Med., 2005, 53, 329–338 CrossRef.
- K. A. Hinds, J. M. Hill, E. M. Shapiro, M. O. Laukkanen, A. C. Silva, C. A. Combs, T. R. Varney, R. S. Balaban, A. P. Koretsky and C. E. Dunbar, Blood, 2003, 102, 867–872 CrossRef CAS.
- C.-L. Tseng, I. L. Shih, L. Stobinski and F.-H. Lin, Biomaterials, 2010, 31, 5427–5435 CrossRef CAS.
- K. B. Ghaghada, M. Ravoori, D. Sabapathy, J. Bankson, V. Kundra and A. Annapragada, PLoS One, 2009, 4, e7628 CrossRef.
- A. C. Silva and N. A. Bock, Schizophr. Bull., 2008, 34, 595–604 Search PubMed.
- A. A. Gilad, P. Walczak, M. T. McMahon, H. B. Na, J. H. Lee, K. An, T. Hyeon, P. C. M.v. Zijl and J. W. M. Bulte, Magn. Reson. Med., 2008, 60, 1–7 CrossRef CAS.
- E. J. Sutton, T. D. Henning, B. J. Pichler, C. Bremer and H. E. Daldrup-Link, Eur. Radiol., 2008, 18, 2021–2032 CrossRef.
- S. Boddington, T. D. Henning, E. J. Sutton and H. E. Daldrup-Link, J. Vis. Exp., 2008, 14, 686 Search PubMed.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS.
- J. Chan, J. P. Menon, R. Mahajan and R. Jandial, Adv. Exp. Med. Biol., 2010, 671, 1–12.
- S. Lee, J. Xie and X. Chen, Biochemistry, 2010, 49, 1364–1376 CrossRef CAS.
- A. Solanki, J. D. Kim and K. B. Lee, Nanomedicine, 2008, 3, 567–578 CrossRef CAS.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS.
- L. A. Bentolila, Y. Ebenstein and S. Weiss, J. Nucl. Med., 2009, 50, 493–496 CrossRef CAS.
- H. Qian, C. Dong, J. Peng, X. Qiu, Y. Xu and J. Ren, J. Phys. Chem. C, 2007, 111, 16852–16857 CrossRef CAS.
- S. J. Lord, H.-l. D. Lee and W. E. Moerner, Anal. Chem., 2010, 82, 2192–2203 CrossRef CAS.
- K. C. Weng, C. O. Noble, B. Papahadjopoulos-Sternberg, F. F. Chen, D. C. Drummond, D. B. Kirpotin, D. H. Wang, Y. K. Hom, B. Hann and J. W. Park, Nano Lett., 2008, 8, 2851–2857 CrossRef CAS.
- L. Prinzen, R. Miserus, A. Dirksen, T. M. Hackeng, N. Deckers, N. J. Bitsch, R. T. A. Megens, K. Douma, J. W. Heemskerk, M. E. Kooi, P. M. Frederik, D. W. Slaaf, M. van Zandvoort and C. P. M. Reutelingsperger, Nano Lett., 2007, 7, 93–100 CrossRef CAS.
- B. Y. S. Kim, W. Jiang, J. Oreopoulos, C. M. Yip, J. T. Rutka and W. C. W. Chan, Nano Lett., 2008, 8, 3887–3892 CrossRef CAS.
- Z. M. Li, P. Huang, R. He, J. Lin, S. Yang, X. J. Zhang, Q. S. Ren and D. X. Cui, Mater. Lett., 2010, 64, 375–378 CrossRef CAS.
- R. C. Han, M. Yu, Q. Zheng, L. J. Wang, Y. K. Hong and Y. L. Sha, Langmuir, 2009, 25, 12250–12255 CrossRef CAS.
- H. Q. Wang, H. L. Zhang, X. Q. Li, J. H. Wang, Z. L. Huang and Y. D. Zhao, J. Biomed. Mater. Res., Part A, 2008, 86a, 833–841 CrossRef CAS.
- M. A. Walling, J. A. Novak and J. R. E. Shepard, Int. J. Mol. Sci., 2009, 10, 441–491 Search PubMed.
- U. Hasegawa, S. I. M. Nomura, S. C. Kaul, T. Hirano and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2005, 331, 917–921 CrossRef CAS.
- W. J. Parak, R. Boudreau, M. Le Gros, D. Gerion, D. Zanchet, C. M. Micheel, S. C. Williams, A. P. Alivisatos and C. Larabell, Adv. Mater., 2002, 14, 882–885 CrossRef CAS.
- W. C. Chan, W. nbsp and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47–52 CrossRef.
- W. J. Kang, M. H. Ko, D. S. Lee and S. Kim, Prot. Clin. Appl., 2009, 3, 1383–1388 CAS.
- W. Mahmoud, A. Sukhanova, V. Oleinikov, Y. P. Rakovich, J. F. Donegan, M. Pluot, J. H. M. Cohen, Y. Volkov and I. Nabiev, Proteomics, 2010, 10, 700–716 CrossRef CAS.
- J. Y. Chen, Y. M. Lee, D. Zhao, N. K. Mak, R. N. S. Wong, W. H. Chan and N. H. Cheung, Photochem. Photobiol., 2010, 86, 431–437 CrossRef CAS.
- J. A. Chen, Y. Pei, Z. W. Chen and J. Y. Cai, Micron, 2010, 41, 198–202 CrossRef CAS.
- W. S. Kuo, Y. C. Ku, H. T. Sei, F. Y. Cheng and C. S. Yeh, J. Chin. Chem. Soc., 2009, 56, 923–934 CAS.
- S. S. Ozturk, F. Selcuk and H. Y. Acar, J. Nanosci. Nanotechnol., 2010, 10, 2479–2488 CrossRef CAS.
- G. P. C. Drummen, Int. J. Mol. Sci., 2010, 11, 154–163 Search PubMed.
- N. Kawashima, K. Nakayama, K. Itoh, T. Itoh, M. Ishikawa and V. Biju, Chem.–Eur. J., 2010, 16, 1186–1192 CrossRef CAS.
- R. J. Errington, M. R. Brown, O. F. Silvestre, K. L. Njoh, S. C. Chappell, I. A. Khan, P. Rees, S. P. Wilks, P. J. Smith and H. D. Summers, Cell Cycle, 2010, 9, 121–130 CAS.
- H. Lee, I. K. Kim and T. G. Park, Bioconjugate Chem., 2010, 21, 289–295 CrossRef CAS.
- Y. H. Hsieh, S. J. Liu, H. W. Chen, Y. K. Lin, K. S. Liang and L. J. Lai, Anal. Bioanal. Chem., 2010, 396, 1135–1141 CrossRef CAS.
- W. C. Schumacher, A. J. Phipps and P. K. Dutta, Adv. Powd. Technol., 2009, 20, 438–446 Search PubMed.
- D. E. Prasuhn, J. B. Blanco-Canosa, G. J. Vora, J. B. Delehanty, K. Susumu, B. C. Mei, P. E. Dawson and I. L. Medintz, ACS Nano, 2010, 4, 267–278 CrossRef CAS.
- J. R. Slotkin, L. Chakrabarti, H. N. Dai, R. S. E. Carney, T. Hirata, B. S. Bregman, G. I. Gallicano, J. G. Corbin and T. F. Haydar, Dev. Dyn., 2007, 236, 3393–3401 CrossRef CAS.
- Y.-W. Noh, Y. T. Lim and B. H. Chung, FASEB J., 2008, 22, 3908–3918 CrossRef CAS.
- L. Yong Taik, et al. , Nanotechnology, 2009, 20, 475102 CrossRef.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010 Search PubMed.
- N. M. Idris, Z. Li, L. Ye, E. K. Wei Sim, R. Mahendran, P. C.-L. Ho and Y. Zhang, Biomaterials, 2009, 30, 5104–5113 CrossRef CAS.
- D. M. Huang, Y. Hung, B. S. Ko, S. C. Hsu, W. H. Chen, C. L. Chien, C. P. Tsai, C. T. Kuo, J. C. Kang, C. S. Yang, C. Y. Mou and Y. C. Chen, FASEB J., 2005, 19, 2014–2016 CAS.
- E. Mooney, P. Dockery, U. Greiser, M. Murphy and V. Barron, Nano Lett., 2008, 8, 2137–2143 CrossRef CAS.
- G. Hilal, et al. , Nanotechnology, 2010, 21, 155101 CrossRef.
- S. K. Basiruddin, A. Saha, N. Pradhan and N. R. Jana, Langmuir, 2010, 26, 7475–7481 CrossRef CAS.
- B. J. Pichler, A. Kolb, T. Nagele and H.-P. Schlemmer, J. Nucl. Med., 2010, 51, 333–336 CrossRef.
- C. Wang and J. Irudayaraj, Small, 2010, 6, 283–289 CrossRef CAS.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- A. Louie, Chem. Rev., 2010, 5, 3146–95 CrossRef.
- R. G. Blasberg, Nucl. Med. Biol., 2003, 30, 879–888 CrossRef.
- H.-Y. Lee, Z. Li, K. Chen, A. R. Hsu, C. Xu, J. Xie, S. Sun and X. Chen, J. Nucl. Med., 2008, 49, 1371–1379 CrossRef CAS.
- J.-s. Choi, J. C. Park, H. Nah, S. Woo, J. Oh, K. M. Kim, G. J. Cheon, Y. Chang, J. Yoo and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 6259–6262 CrossRef CAS.
- L. E. Jennings and N. J. Long, Chem. Commun., 2009, 3511–3524 RSC.
- W. D. Heiss, Eur. J. Neurol., 2009, 16, e4–4 CrossRef.
- M. Nahrendorf, P. Waterman, G. Thurber, K. Groves, M. Rajopadhye, P. Panizzi, B. Marinelli, E. Aikawa, M. J. Pittet, F. K. Swirski and R. Weissleder, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 1444–1451 Search PubMed.
- L. Seulki and C. Xiaoyuan, Mol. Imaging, 2009, 8, 87–100.
- D. W. Townsend, J. Nucl. Med., 2008, 49, 938–955 CrossRef.
- P. Tallury, K. Payton and S. Santra, Nanomedicine, 2008, 3, 579–592 CrossRef CAS.
- J. H. Choi, F. T. Nguyen, P. W. Barone, D. A. Heller, A. E. Moll, D. Patel, S. A. Boppart and M. S. Strano, Nano Lett., 2007, 7, 861–867 CrossRef.
- S. Santra, C. Kaittanis, J. Grimm and J. M. Perez, Small, 2009, 5, 1862–1868 CrossRef CAS.
- B. H. Hasegawa, K. Iwata, K. H. Wong, M. C. Wu, A. J. Da Silva, H. R. Tang, W. C. Barber, A. H. Hwang and A. E. Sakdinawat, Acad. Radiol., 2002, 9, 1305–1321 CrossRef.
- Z. Shi, K. G. Neoh, E. T. Kang, B. Shuter and S.-C. Wang, Contrast Media & Mol. Imaging, 2010, 5, 105–111 Search PubMed.
- K. Vuu, J. Xie, M. A. McDonald, M. Bernardo, F. Hunter, Y. Zhang, K. Li, M. Bednarski and S. Guccione, Bioconjugate Chem., 2005, 16, 995–999 CrossRef CAS.
- D. Patel, A. Kell, B. Simard, J. Deng, B. Xiang, H.-Y. Lin, M. Gruwel and G. Tian, Biomaterials, 2010, 31, 2866–2873 CrossRef CAS.
- C.-W. Lu, Y. Hung, J.-K. Hsiao, M. Yao, T.-H. Chung, Y.-S. Lin, S.-H. Wu, S.-C. Hsu, H.-M. Liu, C.-Y. Mou, C.-S. Yang, D.-M. Huang and Y.-C. Chen, Nano Lett., 2006, 7, 149–154.
- K.-S. Park, J. Tae, B. Choi, Y.-S. Kim, C. Moon, S.-H. Kim, H.-S. Lee, J. Kim, J. Kim, J. Park, J.-H. Lee, J. E. Lee, J.-W. Joh and S. Kim, Nanomed.: Nanotechnol. Biol. Med., 2010, 6, 263–276 CrossRef CAS.
- J. Chung, M. Yamada and P. C. Yang, Curr. Protoc. Stem Cell Biol., 2009,(Chapter 5) Search PubMed Unit 5A 3.
- T. Higuchi, M. Anton, K. Dumler, S. Seidl, J. Pelisek, A. Saraste, A. Welling, F. Hofmann, R. A. J. Oostendorp, B. Gansbacher, S. G. Nekolla, F. M. Bengel, R. M. Botnar and M. Schwaiger, J. Nucl. Med., 2009, 50, 1088–1094 CrossRef CAS.
- B. A. Rzigalinski and J. S. Strobl, Toxicol. Appl. Pharmacol., 2009, 238, 280–288 CrossRef CAS.
- J. Gao, K. Chen, R. Xie, J. Xie, Y. Yan, Z. Cheng, X. Peng and X. Chen, Bioconjugate Chem., 2010, 21, 604–609 CrossRef CAS.
- C. Zhang, T. Liu, J. Gao, Y. Su and C. Shi, Mini-Rev. Med. Chem., 2010, 10, 193–202.
- M. Arsianti, M. Lim, C. P. Marquis and R. Amal, Biomacromolecules, 2010 Search PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 1565–1573 CrossRef CAS.
- V. I. Shubayev, T. R. Pisanic2nd and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS.
- A. K. Gupta, R. R. Naregalkar, V. D. Vaidya and M. Gupta, Nanomedicine, 2007, 2, 23–39 CrossRef CAS.
- C. Sun, J. S. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS.
- J. H. Juang, J. J. Wang, C. R. Shen, C. H. Kuo, Y. W. Chien, H. Y. Kuo, Z. T. Tsai and T. C. Yen, Transplant. Proc., 2010, 42, 2104–2108 CrossRef CAS.
- S. T. Holgate, J. Biomed. Nanotechnol., 2010, 6, 1–19 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |