Hierarchically structured carbon-based composites: Design, synthesis and their application in electrochemical capacitors
C. Z.
Yuan
,
B.
Gao
,
L. F.
Shen
,
S. D.
Yang
,
L.
Hao
,
X. J.
Lu
,
F.
Zhang
,
L. J.
Zhang
and
X. G.
Zhang
*
College of Material Science & Engineering, Nanjing University of Aeronautics & Astronautics, Nanjing, 210016, P.R. China. E-mail: azhangxg@163.com; Fax: +86 025 52112626; Tel: +86 025 52112902
First published on 10th November 2010
Abstract
This feature article provides an overview of the recent research progress on the hierarchically structured carbon-based composites for electrochemical capacitors. The basic principles of electrochemical capacitors, and the design, construction and performance of hierarchically structured carbon-based composites electrode materials with good ions and electron transportation and large specific surface area are discussed. The trend of future development of high-power and large-energy electrochemical capacitors is proposed.
 Changzhou Yuan | Changzhou Yuan received his PhD degree in Materials Science and Engineering under the supervision of Prof. Xiaogang Zhang from Nanjing University of Aeronautics and Astronautics, China, in 2009. He is now an associate professor at Anhui University of Technology, China. His research interests include functional materials and their application in supercapacitors, lithium-ion batteries and fuel cells. |
 Bo Gao | Bo Gao received his MSc in Physical Chemistry from Xinjiang University, China, in 2006. He is currently a PhD student at Materials Science and Engineering, at Nanjing University of Aeronautics and Astronautics, China. His PhD thesis focuses on design and development of carbon nanotubes-based materials and their applications in supercapacitor. |
 Laifa Shen | Laifa Shen received his BSc degree in Applied Chemistry from Qingdao Agriculture University, China, in 2008. He is currently a PhD student at Materials Science and Engineering, at Nanjing University of Aeronautics and Astronautics, China. His PhD thesis focuses on nanomaterials for hybrid battery capacitor. |
 Sudong Yang | Sudong Yang received his MSc in Physical Chemistry from Xinjiang University, China, in 2008. He is currently a PhD student at Materials Science and Engineering, at Nanjing University of Aeronautics and Astronautics, China. His PhD thesis focuses on the synthesis, assembly and characterization of nanostructured graphene materials for applications in catalysis. |
 Liang Hao | Liang Hao received his BSc degree in Applied Chemistry from Nanjing University of Aeronautic and Astronautics, China, in 2007. He is currently a PhD student at Materials Science and Engineering, at Nanjing University of Aeronautics and Astronautics, China. His PhD thesis focuses on the development of electrode materials, in particular, the graphene-based electrode materials for supercapacitors. |
 Xiaogang Zhang | Xiaogang Zhang received his PhD degree in Chemistry from Lanzhou University in 2001. He is now a professor of chemistry at College of Materials Science and Engineering, Nanjing University of Aeronautics and Astronautics. His current research interests include the design and development of nanostructured composites and their applications in energy conversion and storage. |
1. Introduction
Due to concerns over environmental pollution and the depletion of fossil fuels, the development of alternative energy conversion/storage resources with high power and large energy is of particular significance.1,2 Among the various technologies, electrochemical capacitors (ECs) with a capacitance of thousands of Farads, also known as supercapacitors or ultracapacitors, have attracted tremendous attention because of their great advantages including pulse power supply, long cycle life, simple principle and high dynamic of charge propagation.1–5 ECs, as a diverse class of energy-storage devices, incorporate various electroactive materials (high-surface-area carbons, electroactive conducting polymers (CPs), and transition metal oxides, sulfides, nitrides, etc.), electrolytes (conventional aqueous and nonaqueous electrolytes, advanced polymer electrolytes and ionic liquids), and device configurations (symmetric and asymmetric).3–6 Such diversity and design flexibility make it clear why ECs can cover such a broad region on the power vs. energy density plane in the “Ragone plot”, and bridge the critical performance gap between the energy densities of batteries and high power densities offered by conventional dielectric capacitors, and become ideal for the rapid storage and release of energy.1–7 Therefore, ECs are likely to perform an equal significant role to batteries for future energy storage systems. It is their ability to provide the larger energy densities than dielectric capacitors, and higher power densities and longer cycle life than batteries makes ECs widely used in fast-growing portable electronic devices, memory back-up systems for computers, other electronic devices, and particularly, low-emission hybrid electric vehicles (HEVs), where ECs are commonly coupled with primary high-energy batteries or fuel cells to sever as a temporary energy storage device with a high-power capability to store energy.8In general, ECs can be classified into two categories based on the specific energy storage mechanism.3 One is the electrical double layer capacitors (EDLCs) described by the Gouy–Chapman–Stern–Grahame (GCSG) model, where the energy comes from the pure electrostatic charge accumulated at the electrode/electrolyte interfaces, strongly depending on the accessible specific surface area (SSA) of the electroactive carbon materials to the electrolytes and the electronic conductivity themselves. The other is the pseudo-capacitors or redox supercapacitors, in which fast and reversible Faradaic surface or near-surface reactions for energy storage take place. Of note, the two mechanisms commonly function simultaneously depending on the nature of the electrode materials. Moreover, the progress towards ECs can greatly benefit from the continuous development of electroactive materials with appropriate microstructures for fast energy storage.
To obtain high-performance (large specific capacitance (SC) and good electrochemical stability at high rates) ECs, the electroactive materials should meet three requirements, i.e., good electron conductivity, highly accessible electrochemical SSA and efficient mass transport,7–13 which, unfortunately, cannot be performed by single electroactive materials. Therefore, hierarchically structured carbon-based composites have drawn much attention all over the world and have been exploited greatly to address the problems mentioned above.10,14–20 Such unique carbon-based composites can make full use of all nanostructured carbon (e.g. activated carbons (ACs), carbon nanotubes (CNTs), graphene nanosheets (GNS), ordered mesoporous carbons (OMCs), etc.) as a main matrix or support and electroactive materials with Faradaic capacitance, where the two phases are arranged and constructed well with two or more levels in a hierarchical structure. Such composites combine an electric double layer system and a Faradaic pseudo-capacitive system, which can both utilize the fast and reversible Faradaic capacitance coming from the electroactive pseudo-capacitive species and the indefinitely reversible double-layer capacitance at the electrode-electrolyte interfaces. Therefore, hierarchically structured carbon-based composites could be better potential candidates for ECs with larger energy density and higher power property. Only in this way, can the energy density be optimized without deteriorating their high power capability, which is the most critical aspect in the application and development of ECs and determine their ultimate performance. However, it is a great challenge and highly desirable to design and synthesize unique carbon-based composites with hierarchical nanostructures in a controllable and much simpler manner, which can tailor the physical/chemical properties of the electroactive materials to meet the three basic requirements mentioned above for EC applications.
In this feature article, we aim to provide a brief overview of the recently developed progress of the hierarchically structured carbon-based composites for ECs. Moreover, the controllable design and efficient construction of hierarchically structured carbon-based composite electrode materials with good ion and electron transportation, and the dependence of desirable electrochemical performance upon specific microstructures of the novel hierarchical structured carbon-based composites are discussed in detail, which will direct the development of EC technologies.
2. Principle of energy storage in electrochemical capacitors
2.1 The energy storage mechanism of electrochemical capacitors
ECs store energy using either ion adsorption or fast surface redox reactions. Therefore, ECs are divided into two types according to the fundamental mechanisms that govern their capacitance.3 One is the EDLCs, where the capacitance attributed by the accumulation of charges at the electrode/electrolyte interfaces. The typical electrode materials of EDLCs are carboneous materials with high SSA and good electric conductivity, such as, CNTs,9,10,16,20,21 activated carbon fibers (ACFs),22–24 ACs,15,25–27 GNS,11,28–30 OMCs,14,31–33 and so on. The other is redox supercapacitors, where an actual battery-type oxidation–reduction reaction occurs leading to pseudo-capacitance, mainly based on metal oxides (RuO2,34MnO2,33,35Co3O4,36NiO,37etc.), nitrides (for instance, VN38) and sulfides (such as, CoSx, etc.)39 and CPs (polyaniline (PANI),14,28,29,40,41polypyrrole (PPy),42 polythiophenes (PThs),43 poly(3,4-ethylenedioxythiophene) (PEDOT),44etc.).For both a practical and fundamental perspective, ECs are closely related to batteries, and in fact the distinction between these two classes of energy-storage devices has been blurred with the recent advancements in “high-rate” batteries, and with the discovery that many battery materials exhibit capacitor-like electrochemical responses (i.e., “pseudo-capacitance”) when prepared on the nanoscale and in disordered forms.45 In general, however, ECs can be differentiated from high-rate batteries by their operational characteristics: (1) charge–discharge response times that are on the order of seconds; (2) sloping and symmetric charge–discharge profiles; and (3) exceptional cycle life (typically many tens to hundreds of thousands of cycles).6
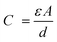 | (1) |
Double layer capacitance for carbon materials in liquid electrolytes is in the range of 5 to 25 μF cm−2, depending on the electrolytes, pore size and pore distribution.49,50 To increase the charge storage, usually, it is necessary to increase the carbon surface area; this is achieved using different synthesis techniques. Generally, SSA can be increased by the development of porosity in the bulk of carbon materials. Unfortunately, there is no simple linear relationship between their SSA and SCs. Indeed, more than the total porosity is created, i.e., control of the pore size as well as pore size distribution (PSD), which has a great influence upon the SCs of carbon materials.2,32,51,52 Traditionally, it was believed that the submicropores of an electrode do not participate in the formation of the electrical double layer (EDL) due to the inaccessiblility of the submicropore surfaces to the large solvated ions. However, Raymundo-Pinero et al.51 recently observed the important contributions of micropores to the overall capacitance and suggested that partial desolvation of hydrated ions occurred, leading to an enhanced SC. Also, Gogotsi et al.2 reported an anomalous SC increase in carbon electrodes with pore sizes less than 1 nm. These interesting results further confirmed an SC contribution from pores with sizes smaller than the solvated ion size, which cannot be fully interpreted by the EDL theory where there would be insufficient room to accommodate both the compact layer and diffuse layer in such confined microporous spaces. Recently, Huang et al.52 have reported an heuristic approach to describing nanoporous carbon-electrode ECs, in which the pore curvature is taken into account and an electric wire-in-cylinder capacitor (EWCC) model is proposed for modeling microporous carbon electrode. And Cheng et al.32 have reported a 3-dimensional hierarchical porous graphitic carbon material with macroporous cores, mesoporous walls and micropores for high rate ECs applications, in their designed hierarchical structure, macropores serves as “ion buffering reservoirs”; the graphitic mesopore walls provide excellent electrical conductivity, capable of overcoming the primary kinetic limits of electrochemical process in porous electrodes, in addition, the presence of micropores can enhance the charge storage.
Therefore, it is significant to increase the SSA by means of making the electroative materials more porous and to optimize their pore size and PSD meanwhile, resulting in the great enhancement of efficient SSA accessible to the electrolytes. Moreover, the existing hierarchical porosity would facilitate the ions diffusion to the carbon surfaces, particularly, at high rates. Even so, one another problem presents itself that the enhancement of the SSA of carbon materials cannot unlimited because that the amount of C atom would decrease greatly in unit area with increasing the SSA, leading to poor conductivity of the carbon materials with larger SSA, which is not favorable for better EDL capacitance.
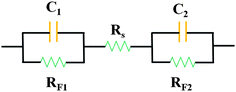
It is convenient to represent the electrical behavior of the electrode interface in terms of an equivalent circuit, the behavior of which would mimic that of the actual interphase with respect to current or potential response to modulation, respectively, of potential or current. Fig. 1 depicts the equivalent circuits for EDL capacitance. The simplest case is, as shown in the dot line pane, that of an EDL capacitance in series with the Rs, which is referred as to the equivalent series resistance (ESR, in ohms), including the resistance of the electrolyte, the intrinsic resistance of active material, the mass transfer resistance of the ions in the matrix, and the contact resistance at the interface of active material and current collector. In the case of same electrolytes and same contact resistance, the intrinsic resistance of electroactive carbon materials plays a great role in keeping good EDL capacitance. There is no charge leakage pathway so that such an interface is referred to as an ideally polarizable electrode. In practical, Faradaic leakage can arise depending on the electrode potential and solution composition, so circuit the shown in the dotted line panel is modified by inclusion of a Faradaic resistance, RF, in parallel across C, which is usually inversely related to the electrode potential or overvoltage according to a Tafel-type equation in exponential form.55
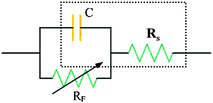 | ||
| Fig. 1 Equivalent circuits for EDL capacitance at an electrode solution interface: capacitor with series resistance (Rs) and potential-dependant Faradaic leakage resistance (RF). | ||
Based on the discussion above, carbon materials with good electrical conductivity and large accessible SSA due to their rich hierarchically porous structures would be the best choice. However, unlimited enhancement of the carbon surface area would be impossible. Moreover, the electrical conductivity of the carbon would decrease greatly with the great enhancement of SSA of carbon materials due to the reducing of carbon atoms in the unit area, which does not facilitate fast and large energy storage. Therefore, dispersing pseudo-capacitive materials with larger SCs onto highly conductive carbon materials with proper porous sizes and PSD to fabricate hierarchical structured composites is also another successful strategy for enhancing the specific energy density of carbon materials without sacrificing their power properties.
As with the representation of double-layer charging behavior, an equivalent circuit representation can be given for charging of a pseudo-capacitance (Cθ) through a Faradaic resistance (RF) and some possible further Faradaic resistance (RD) for discharge (e.g. desorption of an ad-species). This combination shown in Fig. 2 involves Faradaic impedance (Cθ, RF and RD) in parallel with Cdl. Usually, over a certain range of potentials, Cθ can be much larger than Cdl. Of note, Rs related to the electric conductivity of the electroactive materials with pseudo-capacitance is still addressed as another necessary condition for Faradaic reactions. Only if the electrolyte ions and electrons simultaneously access the electroactive sites in time can the Faradaic reactions take place for fast energy storage. Therefore, the electrical conductivity of the electroactive materials is also of significance for Faradaic energy storage. Obviously, the less intrinsic resistance of electroactive materials, the better pseudo-capacitance, which is similar to that of EDL capacitance mentioned above.
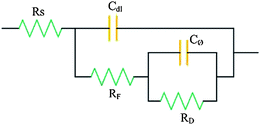 | ||
| Fig. 2 Equivalent circuit for an electrode interface exhibiting Faradaic pseudo-capacitance (Cθ) in a parallel relation through an RF with the EDL capacitance (Cdl). | ||
As mentioned before, pseudo-capacitance is Faradaic in origin, involving fast and reversible redox reactions between the electrolyte and electroactive species on the electrode surface. Thus, two aspects must be resolved carefully. One is the electroactive sites sufficient for Faradaic reactions; the other is sufficient electrolyte ions and electrons participating in Faradaic reactions at high rates simultaneously. As for the former, the larger SSA of electroactive materials with pseudo-capacitance would meet the demand by making the electroactive materials nanosized and/or mesoporous. As for the latter, it would make the electrolyte ions diffuse to and contact the richer electroactive sites in time by creating much more hierarchical (meso- and macro-) porosity of electroactive materials with good electric conductivity. Although pseudo-capacitance can be higher than EDL capacitance, the poor electric conductivity of pure transition metal oxides, such as MnO2 and NiO,10,16,35 and the mechanical degradation of CPs during the Faradaic charging processes,14,40,41,44 hinder their applications as electrode materials. As a result, a potential strategy to overcome these problems is dispersion of the the electroactive materials with good pseudo-capacitance onto the surfaces of carboneous materials with large SSA, hierarchical porosity and good electrical conductivity to synthesize carbon-based composites with a hierarchical structure. The fabrication of composites by using carbon as a support not only increases the effective utilization of electroactive materials with pseudo-capacitance, but also improves the electrical conductivity and mechanical strength of the composite materials, and greatly decreases the amount of electroactive materials with pseudo-capacitance but in the meantime does not sacrifice the good electrochemical performance.
3. Dependence of the electrochemical performance of ECs upon the microstructure of the electroactive materials
ECs are constructed much like a battery in that there are two electrodes immersed in an electrolyte, with an ion permeable separator located between the electrodes. In such a device, each electrode-electrolyte interface represents two capacitors so that the complete cell can be considered as two capacitors in series. For an EC, the net cell capacitance will therefore be calculated according to the following equation: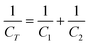 | (2) |
Therefore, any degree of difference between the positive and negative pair of electrodes in a single cell will result in the overall capacitance of the device, which is determined by the electrode with the smaller SC. And if they are approximately equal, it is seen that the overall C of any “symmetric” capacitor is only half that of the individual capacitance (the SC of a single electrode reported in the literature is based on a three-electrode cell configuration) of each electrode in the cell. Notably, this is a quite general result of combining capacitive elements in series and has to be taken into account in device design to optimize performance specifications and delivered efficiency.
The basic operation of a single-cell EC consisting two electrodes also can be illustrated in a simple equivalent circuit representation, as shown in Fig. 3. The whole performance of an EC depends upon the SCs of each electrode, can be found to be related to the microstructures (SSA, porosity, and electric conductivity, etc.) of the electrodes, assuming the same electrolytes, as discussed above.
 | ||
| Fig. 3 A simple equivalent circuit representation demonstrating the basic operation of a single EC. | ||
The performance of ECs is commonly evaluated on the basis of the following criteria: (1) a power density substantially greater than batteries with acceptably high energy densities (>10 Wh kg−1); (2) an excellent cyclability (more than 100 times that of batteries); (3) fast charge-discharge processes within seconds. The maximum energy storage (E) and power delivered (P) for a single EC are given in eqn (3) and (4):
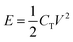 | (3) |
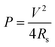 | (4) |
Based on the discussion above, it is evident that the whole electrochemical performance of an EC mainly depends upon both the two electrodes in the given electrolytes. Specifically, the three aspects are of being noted, as shown in Scheme 1. One is the electric conductivity, which can be enhanced by decreasing the size of the electroactive material and/or adding carbon with good electric conductivity (by many ways); another is the larger SSA, which can be obtained by synthesizing nanosized or/and micro- and meso- porous electroactive materials; the final is convenient ion transportation, which can be performed by prepared hierarchical (meso- and macro-) porous electroactive materials, particularly, the oriented arrays with ordered pores for fast ions transportation at high rates.
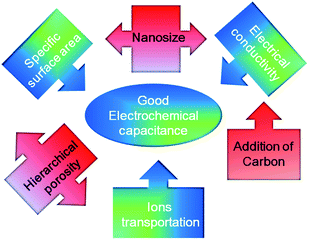 | ||
| Scheme 1 Dependence of the electrochemical performance of ECs upon the microstructure of the electroactive materials. | ||
4. Design, synthesis and performance of hierarchically structured carbon-based composites
According to the above discussion, hierarchically structured carbon-based composites would be better candidates for EC applications, in which pseudo-active materials are incorporated into carboneous materials with a defined SSA and PSD to optimize the overall capacitance and conductivity of the electrodes without destroying the electrochemical stability and high rate property, desirable for their practical application. It is due to their extraordinary properties, including high electrical conductivity, unique geometrical structure and high SSA, that it is a currently hot research area how to precisely control the morphologies, PSD, composition and crystal structure of the composites at the nanoscale to meet the three basic requirements for ECs application mentioned above. Areas specifically highlighted in the following section present recent research work in the synthesis strategies, processing, control of multi-scale structures and electrochemical properties of carbon (including ACs, OMCs, CNTs (arrays), GNS and other carbon forms)-based composites with hierarchical structure.4.1 Activated carbons-based composites
ACs are widely used as ECs electrode materials due to their large SSA, relatively good electrical properties, excellent thermal stability and low cost. Depending on the carbon precursors as well as the activation methods used, ACs possessing various physicochemical properties with SSA as high as 3000 m2 g−1 have been produced and their electrochemical properties have been studied.55–58 In general, the SC of ACs is higher in aqueous electrolytes (ranging from 100 F g−1 to 300 F g−1) than in organic electrolytes (less than 150 F g−1). The larger values in aqueous electrolytes are essentially justified by a smaller size of solvated ions and a higher dielectric constant than in organic media. However, organic electrolytes are generally preferred for applications, due to their high potential window, which allows more energy to be stored than in aqueous solution. Besides, some other aspects of the ACs materials, such as, PSD, electrical conductivity and surface functionality can also influence their electrochemical performance to a great extent.In addition, AC-based composite for ECs have been also investigated. By addition of metal oxides or CPs on the ACs used for EDLC electrodes, SC can be enhanced. Zhang et al.59 reported that hydrous ruthenium oxide/AC electrode (RuO2·xH2O/AC) with a 35 wt% ruthenium oxide loading delivered an SC of 350 F g−1, much larger than that of 243 F g−1 for pure AC. In ref. 60, for only 3.2 wt% ruthenium oxide in the composite electrode, an increase in the SC of ca. 25% to a value of 324 F g−1 was reported. Vijayamohanan et al.61 reported that the composite electrode showed an enhanced SC of 250 F g−1 in 1 M H2SO4 with 9 wt% ruthenium incorporated. Other experiments with RuO2/AC composites used as positive electrodes in ECs also indicated the increase of SC.62 However, RuO2 is a very expensive material in limited supply. Much work has been focused on replacing RuO2 with other cheap metal oxides that also exhibit good electrochemical behaviors.
NiO is one of the most studied materials as a low-cost alternative to RuO2. Yuan et al.63 reported that some nanometre-scale amorphous particles of nickel oxide loading onto AC can enhance the SC and the energy density of AC-based ECs. Its SC increased 10.84%, from 175.40 to 194.01 F g−1. Similar enhanced SC also took place in manganese oxide/AC composite electrodes.64Ni(OH)2/AC composite electrode has been extensively studied due to its better electrochemical reversibility, higher proton diffusion rate and a better discharge property.65 The unique composite used in ECs provided a significant increase of SC from 255 to 314 F g−1 when the loading amount of Ni(OH)2 was 6 wt% and also good electrochemical performance and high charge–discharge properties.66 Other materials used for AC composite electrodes67,68 have been found to increase the SC of electroactive materials. In ref. 67fullerene-activated carbon composite electrodes were prepared and their charge/discharge characteristics were studied for use in a high power EDLC. A higher SC of 172 F g−1 was obtained at 50 mA cm−2 on a 1 wt% C60-loaded electrode with ultrasonic treatment.
In addition, AC-based CPs composites, such as PANI/AC, PPy/AC and PThs/AC can also display larger SCs.69,70 Li and co-workers70 found that the SC of the PANI/AC composite electrode was 587 F g−1, which was much higher than that of the pristine AC (ca. 140 F g−1), owing to the Faradaic reaction of PANI with the electrolyte. Doped CPs can also display high SC, but the relatively poor stability of the organic materials has so far limited their applications greatly.
The specific pseudo-capacitance exceeds that of carbon materials, justifying the interest in these systems. However, pseudo-capacitors, like batteries, often suffer from a lack of stability during cycling. Therefore, we should design and construct other carbon based materials. In traditional high-surface-area ACs, the diffusion pathway for the ions is tortuous (Fig. 4a). At high current density, the inner surface accessibility is limited and results in a high resistance, causing a loss of SC for thick samples. Therefore, it is necessary to design other carbons with narrow PSD (accessible to the electrolyte ions diffusion) with an interconnected pore structure and short pore length together with controlled surface chemistry, for example, OMC (Fig. 4b) would be beneficial for enhancing the energy density of ECs, without deteriorating their high power density and cycle life. The key drawbacks associated with ACs materials are: (a) slow mass transport of molecules because of space confinement imposed by small pore sizes;71 and (b) low conductivity arising from of the presence of enormous surface functional groups and defects.72
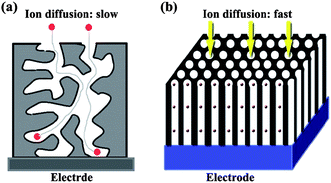 | ||
| Fig. 4 Illustration of diffusion of electrolyte ions in AC (a) and OMC (b). | ||
4.2 Ordered mesoporous carbon-based composites
OMCs, as a new kind of porous carbon materials, have been used extensively as electrode materials for batteries, fuel cells and sensors because of their unique physical and chemical properties. The synthesis of them are mainly based on the templating techniques, i.e., the hard-template and soft-template method. The hard-template synthesis method is defined as replication synthesis with presynthesized hard templates through impregnation, carbonization and template removal. The soft-template synthesis method refers to self-assembly using soft templates through cocondensation and carbonization. Both two methods can offer an effective way to produce nanostructured carbons with large SSA, well-controlled narrow PSD, ordered pore structures and an interconnected pore network, and thus make them promising candidates for EC electrode materials.Since Hyeon et al.73 first reported that the OMC exhibits excellent electrochemical performances compared to the commercially available carbon, OMCs have received attention as electrode materials for EDLCs. Zhou et al.74 have investigated the capacitive properties of the self-ordered mesoporous carbons (CMK-3), from a template of SBA-15 used sucrose as the carbon source. Centeno et al.75 have discovered that the design of the PSD of the electroactive materials is required to suit the size of electrolyte ions and enhance the performance at high rate. Xing et al.76 have reported superior EDLCs properties of OMC by varying the ordered pore symmetries and mesopore structure. Compared to a commercially used AC electrode, Maxsorb, these OMC with large mesopores and especially with 2-D pore symmetry, have superior capacitive behavior, power output and high-frequency performance. This can be attributed to the unique structure of their mesopore network, which is more favorable for fast ionic transportation than the pore networks in disordered microporous carbons. Yamada et al.77 have synthesized ordered porous carbons containing meso/macro/micropores with large SSA. They found that the OMC having well controlled hierarchical porous structures would be favorable for designing high performance EDLCs electrodes. The investigation on pore-dependent capacitance properties have revealed that the micropores adjacent to the open mouths of pores are effective in charge storage, and larger pores that are interconnected are important for smooth electrolyte transportation. Furthermore, it is worth mentioning that Gogotsi et al.78,79 have investigated the effect of the pore size on SC. They have detected an anomalous increase in capacitance at pore sizes of the carbon materials less than 1 nanometre (Fig. 5). This results challenge the long-held axiom that pores smaller than the size of solvated electrolyte ions are incapable of contributing to charge storage.
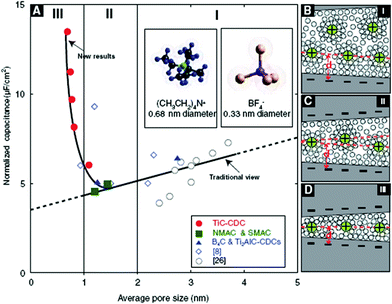 | ||
| Fig. 5 (A) Plot of SC normalized by BET SSA for the carbons in this study and in two other studies with identical electrolytes. The normalized capacitance decreased with decreasing pore size until a critical value was reached, unlike the traditional view which assumed that capacitance continually decreased. It would be expected that as the pore size becomes large enough to accommodate diffuse charge layers, the capacitance would approach a constant value. (B to D) Drawings of solvated ions residing in pores with distance between adjacent pore walls (B) greater than 2 nm, (C) between 1 and 2 nm, and (D) less than 1 nm illustrate this behavior schematically. Reproduced with permission from ref. 78. Copyright Science Publishing Group, 2006. | ||
In addition to directly acting as electrode materials for EDLCs, OMC also can be used as supporting materials for pseudo-capacitive materials loading owing to its high conductivity, narrow distribution of mesopore sizes, and large SSA as well as the ability to form a three dimensional conducting network. Xing et al.80 synthesized a PANI/OMC composite for ECs by an in situ chemical polymerization method. Structure characterization shows that the PANI is well coated on the pore surface of OMC. SC of PANI/OMC reaches as high as 602.5 F g−1 in charge–discharge tests, which is much higher than that of OMC, due to the incorporation of PANI onto the pore surface of OMC. Song et al.81 have investigated in detail the influence of the compounding process between OMC and PANI on the supercapacitive performance of PANI/OMC composite. They prepared PANI/OMC composites via different processes, involving the in situpolymerization of aniline in the presence of OMC or its precursor and the direct physical mixing method. It has been found that the composite prepared by using OMC as a starting material possesses not only a high SC (747 F g−1) but also a good rate capability. The reason can be ascribed to the high degree of dispersion of PANI molecules and double fixing effects of the surface and mesopore of OMC on PANI, which made more PANI molecules available for Faradaic reaction and OMC a stronger support for the maintenance of electrical conductivity and mechanical strength of PANI. Xia et al.14 prepared ordered whiskerlike PANI on the surface of OMC through an appropriate synthesis process. Loosely packed nanometre-scale PANI whiskers create electrochemical accessibility for electrolyte ions and reduce the distance within the PANI bulk that ions must be transported during the charging/discharging process. The high conductivity of OMC greatly reduces the energy loss and power loss due to IR loss at high charge–discharge current density. The SC of the PANI/OMC composite is as high as 900 F g−1 at a charge–discharge current density of 0.5 A g−1 and the capacitance retention of this composite is higher than 85% when the current density increases from 0.5 A g−1 to 5 A g−1. Kong et al.82 synthesized three kinds of hierarchically porous Ni(OH)2/CMK-3 composites by a facile chemical precipitation method. It was found that the special structure of the composites greatly improve the property of Ni(OH)2. Firstly, the macro/mesopores formed by interconnected nickel hydroxide nanoflakes would provide fast diffusion channels for electrolyte and act as ion-buffering reservoirs to reduce the diffusion distances to the interior surfaces. Second, the small mesoporous structure, mainly originating from the CMK-3 mesopore wall, can provide low-resistant pathways for the ions through the porous structure, as well as a shorter diffusion route because of the ordered mesoporous channels. In addition, the micropores located within the mesopore wall are expected to be most efficient in a double-layered formation. As a result, their BET SSA were 322, 231, 288 m2 g−1, respectively, which are much higher than that of the pure Ni(OH)2 (99 m2 g−1). The as-prepared composites possess a highest SC of 2570 F g−1 at a current density of 5 mA cm−2 and offer a better high-rate discharge ability. Even though under the large discharge current density of 50 mA cm−2, nearly 86% of the initial capacitance can be reached. Under annealing at a certain temperature, the composite could change into NiO/CMK-3, which is probably promising electrode material. Cao and co-workers83 have successfully prepared NiO/CMK composites by carbonization of sucrose in the presence of nickel acetate inside an SBA-15 template. The nickel oxide nanoparticles (NPs) are formed during carbonization inside the mesoporous pores of SBA-15. The PSD of the obtained material is mainly located at 3–5 nm with a BET surface area of 1262 m2 g−1. Owing to the unique structure incorporation of CMK, the ions can diffuse into the electroactive surface more conveniently. Besides, the different pore arrangement and the presence of a more secondary microporosity play an important role. The electrochemical results show that the NiO/CMK electrode possessed an SC of 230 F g−1. Shi et al.84 have synthesized novel structured MnO2/mesoporous carbon composites with MnO2 NPs embedded into the mesoporous carbon wall of CMK-3 materials. This structure is important for the mass-transfer process in the electrode reaction. Consequently, these materials show good pseudo-capacitive properties and high electrochemical stability, which have an SC of over 200 F g−1 for the composite and 600 F g−1 for MnO2, and maintain 92% of the original value after 1000 cycles. Li et al.85 prepared RuO2/CMK-3 composites by the impregnation of CMK-3 carbon with RuCl3 solution, which reached a high SC of 633 F g−1 due to the high SSA of CMK-3 mesoporous carbon and the pseudo-capacitance of amorphous RuO2. Furthermore, the studies also showed that, compared with microporous carbon, the mesoporous carbon is a better support, because the deposited RuO2·xH2O NPs could block the carbon micropores.86
It is necessary to point out that the different methods of preparation OMC have remarkable effect on the characteristics of OMC. Three-dimensional OMC (3DOMC) has a 3D network path for electronic conduction and high porosity of more than conventional OMC.87 Dokko et al.88 synthesized a composite electrode consisting of bimodal porous carbon and PANI by the electropolymerization of aniline within the macropores of bimodal porous carbon. The electrochemical properties of the composite were characterized in a mixed solution of ethylene carbonate and diethyl carbonate containing 1 M LiPF6. The discharge capacity of the PANI/3DOMC composite electrode is 111 mAh g−1 in the potential range of 2.0–4.0 V (vs. Li/Li+), which corresponds to a volumetric discharge capacity of 53 mAh cm−3. The composite also shows good rate capability, and the discharge capacity at a high current density of 6 A g−1 is as high as 81 mAh g−1.
In addition, it is generally accepted that the interconnected, hierarchical porous structure is essential for the successful synthesis of porous carbon materials for ECs electrodes because the presence of mesopores can improve the power performance at high current densities and accelerate the kinetic process of the ion diffusion in the electrodes, whereas micropores that are accessible to the electrolyte ions are essential for high energy storage. Cheng and co-workers32 synthesized a 3-dimensional (3D) hierarchical porous graphitic carbon (HPGC) material with macroporous cores, mesoporous walls and micropores. The four unique characteristics of their HPGC (namely, macroporous cores as ion-buffering reservoirs, mesoporous walls with smaller ion-transport resistance, micropores for charge accommodation, and a localized graphitic structure for enhanced electric conductivity) make it a suitable support for transition metal oxides as electrode material for high-performance ECs. The schematic shown in Fig. 6 illustrates the 3D hierarchical porous structure of transition metal oxide/HPGC composite as imagined.
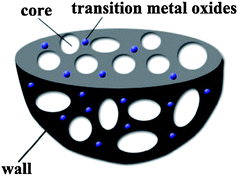 | ||
| Fig. 6 The 3D hierarchical porous structure of transition metal oxide/HPGC composites. | ||
4.3 Carbon nanotube-based composites
CNTs have been widely studied since they were discovered in 199189 and attracted extensive attention. CNTs are formed when a graphite sheet is curled up into cylinders, including single-walled CNTs (SWCNTs) and multi-walled CNTs (MWCNTs).91 CNTs have a novel structure, a narrow size distribution in the nanometre range, highly accessible surface area, low resistivity, and high stability.89–93 These features exactly meet the characteristics that ECs require; thereby both SWCNTs and MWCNTs have been extensively studied for either EDLC electrodes or as good substrates for pseudo-capacitive materials.94–100Unfortunately, owing to their inherent strong van der Waals interactions and poor interface compatibility with the matrix, the CNTs hold together as large bundles and present intrinsic chemical stability, which causes inferior electrochemical properties. So, the CNTs should be pretreated or modified before their practical application. The main approaches for the modification of these CNTs used for electrode or substrate can be grouped into two categories: (a) the covalent attachment of chemical groups through reactions onto the π-conjugated skeleton of CNTs; (b) the noncovalent adsorption or wrapping of various functional molecules.
For the first approach, the most common method is oxidized treatment with strong oxidants including hydrogen peroxide, potassium permanganate, potassium hydroxide, and nitric and/or sulfuric acid. For example, the use of functionalized CNTs (f-CNTs) for EDLCs was first reported by Niu et al.,94 where CNTs were first pretreated in nitric acid. These f-CNTs were found to possess an SSA of 430 m2 g−1. A gravimetric SC of 102 F g−1 and an energy density of 0.5 Wh kg−1 could be obtained at 1 Hz in an acidic electrolyte. In these reports, the strong oxidation process can create a large number of hydrophilic carboxyl groups on the CNTs’ surface.97 These functional groups have been found not only to improve the dispersity of MWCNTs, but also to produce the additional pseudo-capacitance. So both the Faradaic and non-Faradaic processes were involved in the CNTs-based EDLCs, causing the value of SCs increased significantly.101 However, when CNTs were oxidized with those extremely aggressive agents, this process, inevitably, causes some damage or structural change on the CNTs’ surfaces by disrupting their graphene sheet structure. This structure changes result in the decrease of electrical conductivity, which is negative in terms of the application for EDLCs. Hence, the balance between hydrophilicity and conductivity is therefore a key factor when designing improved electrode materials for EDLCs, and oxidized process must be optimized first.102–105
As a whole, the application of CNTs in ECs remains a challenge because of its high cost and highly variable purity and quality as well as relative low SC although its reported electrochemical properties have achieved some progress. The demand to develop new alternate electrode materials that obtain a higher energy density but maintain a high power density for ECs is still very urgent. Therefore, hybrid composites of CNTs and pseudo-capacitive materials consisting of a nanotubular backbone coated by an active phase with pseudo-capacitive properties, such as metal oxide/CNTs and CPs/CNTs composites, represent an important breakthrough for developing a new generation of ECs based on the following basic reasons:106–111 (1) the synergy between EDLCs and pseudo-capacitors can achieve complementary advantages of two types of ECs materials; (2) the open mesoporous network formed by the entanglement of nanotubes allows the ions to diffuse easily to the active surface of the composite components, which lowers the ESR; and (3) since the nanotubular materials are characterized by a high resilience, the composite electrodes can easily adapt to the volumetric changes during charge and discharge, which improves drastically the cycling performance. Furthermore, besides improving the capacity, a critical issue here is that the organic groups, defects or polymers which functionalized on the surface of CNTs is also helpful to the formation of uniform morphologies or the improvement in the electrochemical performance. Park et al.112 found that the electrostatic charge storage as well as pseudofaradaic reactions of RuO2 NPs can be affected by the surface functionalization of CNTs due to the increased hydrophilicity. Such hydrophilicity enables easy access of the solvated ions to the electrode/electrolyte interface, which increases the Faradaic reaction site number of RuO2 NPs and leads to higher SCs. The SC of RuO2/hydrophilic CNTs nanocomposites was found to be ca. 120 F g−1 (13 wt% RuO2 loading) while the SC of RuO2/pristine CNTs nanocomposites was only ca. 70 F g−1 under same RuO2 mass loading. Kim et al.113,114 reported that the highly dispersed hydrated RuO2 NPs can be obtained on carboxylated CNTs by preventing agglomeration among RuO2 NPs through bond formation between the RuO2 and the surface carboxyl groups of the CNTs (Fig. 7). The highly dispersed RuO2 NPs on CNTs show an increased SCs, which can be attributed to the amorphous and porous structural ruthenium oxide with a small size that provides a large SSA and forms conduction paths for protons to access easily even the inner part of the RuO2. A prominently enhanced capacitive performance was also observed in well-dispersed RuO2 NPs on nitrogen-containing CNTs.115,116 The function of nitrogen amalgamation is to create preferential sites on CNTs with lower interfacial energy for attachment of RuO2 NPs. This crucial phenomenon leads to a significant improvement in the overall SCs. Xia et al.117 reported that the hydrophilic CNTs can reduce the aggregation of Ni(OH)2 NPs, inducing a good distribution of the nanosized Ni(OH)2 NPs on the cross-linked, netlike structure CNTs. The rate capability and utilization of Ni(OH)2 were greatly improved, and the composite electrode resistance was reduced. A specific energy density of 32 Wh kg−1 at a specific power density of 1500 W kg−1 was obtained in the hybrid ECs. Zhang et al.107 studies the pseudo-capacitive performances of Co–Al layered double hydroxide (LDH) mixing with hydrophilic MWCNTs. It is found that either SC (342.4 F g−1) or long-term working performance of this composite at high current density is superior to pure LDH electrode (192 F g−1) due to the network of MWCNTs attached to the surface of LDH, which improves interconnection of Co–Al LDH particles and decreases contract resistance. In addition to hydrophilic carboxylated CNTs, there are many other functionalized CNTs used for enhancing electrochemical performance of the composites. For instance, core–shell structural RuO2·xH2O/CNTs composite can be obtained by using covalent benzenesulfonic functionalized CNTs as substrate. Besides enhancing the dispersity of CNTs in water, benzenesulfonic groups can simultaneously act as the anchoring site to tether Ru3+ ion precursors onto its surface. The maximum SC of 1180 F g−1 for RuO2·xH2O was obtained from this composite with 32 wt% RuO2·xH2O, which was much higher than that of just 840 F g−1 for the RuO2·xH2O/pristine CNTs.118 Ultrasonicated CNTs119 have also been proven to be an effective substrate for chemical deposition of pseudo-capacitive material. Because the ultrasonic cavitation destroys the outer wall of CNTs, a lot of nicks were formed which then acted as nucleation sites to favor NiO coating uniformly onto CNTs. The NiO/CNTs electrode presents a maximum SC of 523 F g−1, which can be attributed to the three-dimensionally interconnected nanotubular structure with a thin film of electroactive materials.
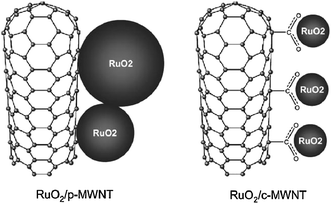 | ||
| Fig. 7 High dispersion of RuO2 due to the surface carboxylated groups of MWCNTs in the nanocomposites. Reproduced with permission from ref. 113. Copyright The Royal Society of Chemistry, 2005. | ||
On the other hand, the composites based on CNTs functionalized by CPs,120,121 such as PPy and PANI, have attracted much attention recently because the CPs here are bifunctional both for functionalizing CNTs and producing the pseudo-capacitance.122–124 Furthermore, because the entangled mesoporous network of nanotubes in the composite can adapt to the volume change, the drawbacks of CPs such as the shrinkage breaking, and cracks of the polymer chain appearing in long-term cycles can be avoided, and hence a more stable capacitance with cycling can be obtained. Frackowiak et al.122 found that a high SC value could be achieved by hybridizing the CPs (PANI and PPy) with CNTs. Gupta et al.123 presented that the SC of PANI/SWCNTs composite increased as the amount of the deposited PANI onto SWCNTs increases, where the PANI was wrapped around SWCNTs. The highest SC, specific power, and specific energy values of 485 F g−1, 2250 W kg−1 and 228 Wh kg−1 were observed for 73 wt% PANI deposited onto SWCNTs, respectively. And the PANI/SWCNTs composites showed long cyclic stability. Li et al.124 reported that PANI/SWCNTs composite films with good uniformity and dispersion were synthesized by electrochemical polymerization of aniline containing hydrophilic SWCNTs. As they adhere strongly to aniline by the formation of a charge-transfer complex rather than by weak van der Waal's interactions, SWCNTs can act as an effective dopant, which facilitates charge transfer between the two components from a localized to a more delocalized band structure and promotes the portonation of PANI, resulting in enhanced electroactivity and increased electrical conductivity. Zhang et al.41 reported a core–shell structural nanocomposite of benzenesulfonic MWCNTs doped PANIvia an in situpolymerization method. For this composite, as a result of the existence of a large number of benzenesulfonic acid groups, MWCNTs can serve as a large molecular dopant, which is integrated and essentially locked to the PANI chains. This polyemeraldine salt with the polyanion is extremely stable and ensures the better desired electrochemical capacity and cyclability once it is formed.
However, it must be noted that covalent functionalization or other modification methods mentioned above have a similar effect as the oxidizing process. Those processes lead to a decrease of electrical conductivity, which is negative for the power characteristics of the composite. Therefore, noncovalent functionalization of CNTs is becoming particularly attractive because it offers the possibility of attaching chemical handles without affecting the electronic network of the tubes. The noncovalent interaction is based on van der Waals forces or π–π stacking, and it is controlled by thermodynamics. Recently, poly(styrene sulfonate) (PSS) has been considered to be an effective noncovalent dispersant that involves the non-covalent functionalization of CNTs with negatively charged PSS, and creates active sites on the CNTs surfaces.125 For instance, Zhang et al.20 reported that RuO2·xH2O nanodots can be highly dispersed onto the surfaces of PSS functionalized CNTs (PSS-CNTs) via a polymer-assisted technique under mild hydrothermal conditions. PSS served as a bifunctional molecule both for solubilizing and dispersing MWCNTs into aqueous solution and for tethering Ru3+ to facilitate the subsequent uniform formation of RuO2·xH2O nanodots on their surfaces (Fig. 8). The maximum SC of 1474 F g−1 and electrochemical utilization of 71% for the Ru species were obtained from RuO2·xH2O/PSS-CNTs nanocomposite with 10 wt% RuO2·xH2O. They also employ PSS-CNTs as substrate to synthesize the ordered mesoporous NiO/PSS-CNTs with good electrochemical performance. PSS-CNTs,10 as a good three-dimensional conducting network, not only enhanced the conductivity of the composite, but facilitated the soaking of the electrolyte into particles, maintaining its sustentation, and created a much more porous channel for electrolyte ions to transport and electrochemically access even more electroactive sites of the ordered mesoporous NiO for energy storage at larger current densities. Electrochemical data demonstrated the unique composite (ca. 48 wt% NiO) could deliver a large energy density and high power property, and SC of 439 F g−1 could be obtained at 6 A g−1. Other workers also successfully used the PSS-CNTs as a substrate to synthesize composites with superior electrochemical capacitive behavior such as PSS-CNTs/PEDOT/MnO2,126PPy/PSS-CNTs127 and PEDOT/PSS-CNTs.44
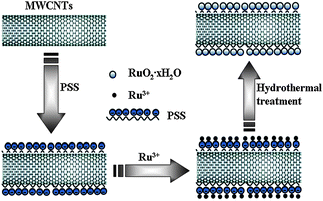 | ||
| Fig. 8 A schematic representation of the formation process of RuO2·xH2O/CNTs nanocomposites. | ||
Recently, with the vigorous developments in soft portable electronics and digital devices, flexible ECs have attracted more attention since they combine the advantages of high power of conventional capacitors and the high specific energy of ECs. In this type of electrodes, CNTs not only act as a highly conductive, flexible, and active substrate, but also yield significantly improved electrochemical performance of supercapacitor electrodesvia enhancement in the electrode/electrolyte interface areas and decrease in the cation diffusion length within active materials. Cui et al.128 reported a carbon nanotube thin film-based ECs fabricated with printing methods (Meyer rod coating or ink-jet printing), where electrodes and separators are integrated into single sheets of commercial paper. The device structure is extremely simple and flexible, with SWNT films behaving as both the electrodes and the current collectors and paper as both the substrates and separator (Fig. 9). In addition, the method for fabricating such a device is scalable and could be accomplished through roll-to-roll methods. A SC of 33 F g−1 at a high specific power of 250 Wh kg−1 is achieved with an organic electrolyte. Wallace et al.129 also reports a simple, scalable, and cost-effective process for the growth of 3D CNTs entangled networks integrated within a highly conducting nongraphitized carbon layer on various conductive and non-conductive substrates. These networks possess a high SSA and superior electrochemical properties. The “freestanding” carbon nanotube/carbon layer (CNT/CL) paper electrodes so obtained are lightweight, flexible, exhibit high conductivity, and can be fabricated into a testing coin cell without using a metal substrate or binder. When employed in a Li-ion battery, the CNT/CL paper electrode shows a significant and fully reversible capacity of 572 mAh g−1 after 100 cycles, which is also suitable for use in other electrochemical devices, such as ECs. Chou et al.130 have prepared totally flexible MnO2 nanowire/CNTs composite paper by electrochemical deposition of MnO2 nanowires into/onto CNTs paper through a cyclic voltammogram technique. Electrochemical measurements showed that the MnO2 nanowires/CNT paper electrode displayed SCs as high as 167.5 F g−1 at a current density of 77 mA g−1. Chen et al.131 have successfully fabricated flexible electrode by direct growth of PANI nanowiresviaelectrochemical deposition onto carbon cloth surfaces and also demonstrated excellent capacitive performance in both gravimetric as well as area-normalized capacitance values for these PANI nanowires/carbon cloth electrodes. According to the galvanostatic charge/discharge analysis, the gravimetric capacitance of 1079 F g−1 was obtained at a specific energy of 100.9 Wh kg−1 and a specific power of 12.1 kW kg−1. Significantly, an extremely high magnitude of the area-normalized capacitance (1.8 F cm−2) can be achieved in this novel nanocomposite wherein the architecture is favorable for effective cation diffusion.
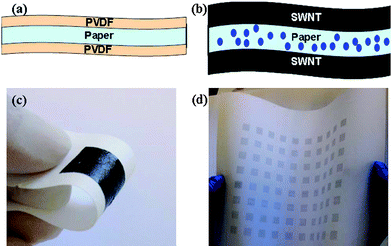 | ||
| Fig. 9 (a) Schematic of Xerox brand printer paper treatment with PVDF; this treatment is used to block the micron-sized pores to avoid short circuiting because of SWNT penetration. (b) Paper supercapacitor structure with SWNT film printed on both sides of the treated Xerox paper. The SWNT film is either Meyer rod-coated or ink-jet printed. (c) A photo of a Meyer rod-coated supercapacitor on Xerox paper. (d) A photo of ink-jet printed supercapacitor on Xerox paper. Reproduced with permission from ref. 128. Copyright American Institute of Physics, 2007. | ||
Commonly, the CNTs are palletized in a paste electrode for evaluating their electrochemical performance. In this case, the MWCNTs will be randomly entangled and cross-linked, thereby forming an entangled CNTs (ECNT) electrode (Fig. 10b) with irregular pore structures. This would lead to a great loss of the inherent advantages of CNTs and eventually limit the improvement of EDLCs performance. Compared to the entangled CNTs, aligned carbon nanotube arrays (CNTA) possess more regular pore structures and conductive paths, thus, they have higher effective SSA and lower ion diffusion resistances, which have a profound influence on their electrochemical properties and their applications in EDLCs.132,133 For instance, Hata et al.12 prepared an aligned high-densely packed SWNT-based CNTAs electrode with higher ion conductivity, which exhibits maximum power density of 43.3 kW kg−1, higher than that of ECNT-based ECs previously reported by Niu et al.94 (approximately 8 kW kg−1). Pan et al. reported a new form of thin films formed by MWCNTs exhibiting high packing density and local alignment, which were fabricated using a highly concentrated colloidal suspension of carbon nanotubes. EDLC built from these film electrodes exhibited nearly ideal cyclic voltammograms close to rectangular and high energy density (30 kW kg−1) even at a high scan rate of 1 V s−1.134,135 In order to further increase the electrochemical performance of the CNTA electrode, a general strategy is growing pseudo-capacitive materials on CNTA electrodes to fabricate hybrid nanostructured electrodes with superior capacitive properties. These hybrid nanostructured electrodes can be divided into two categories: metal oxide/CNTA electrodes (Fig. 10c) and CPs/CNTA electrodes (Fig. 10d). These hybrid nanostructures electrodes have several advantages. First, the pseudo-capacitive NPs is contacted directly with carbon nanotube arrays, thus this superior conducting network allows for efficient charge transport and enhances the electronic conductivity of the composite significantly.136 Secondly, the regular pore structures enhance the ionic conductivity of the CNTA-based hybrid nanostructured electrodes greatly. The well-defined transport channel results in the lowest ion diffusion resistances. Thirdly, the NPs or polymer films well dispersed or coated onto CNTA have the high SSA and the nanometre size, which increases the effective contact area between electrolyte and active material, ensuring a high utilization of electrode materials, and then a high SC.137 For example, Cao et al.16 have reported the synthesis of manganese oxide nanoflower/CNTA composite electrodes with hierarchical porous structure, large SSA and superior conductivity. The electrochemical studies have shown that the manganese oxide/CNTA composite possessed SC of 101 F g−1 at high current density of 77 A g−1 and good cyclability, which is better than the manganese oxide–ECNT composites. They137 also reported the nanobrush-like PANI/CNTA composite electrode, by a combination of a CNTA framework and electrodeposition technique. The electrochemical studies have shown that a very high SC of 1030 F g−1 was obtained for the PANI/CNTA composite as well as high rate capability (95% capacity retention at 118 A g−1) and good stability (only 5.5% loss up to 5000 cycles test).
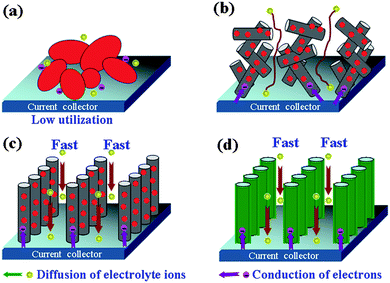 | ||
| Fig. 10 Schematic representation of transportation of electrons and electrolyte ions in (a) metal oxide electrode, (b) metal oxide/ECNTs compositions electrodes, (c) metal oxide/CNTA compositions electrodes, (d) CPs/CNTA composite electrodes. | ||
4.4 Graphene-based composites
Recently, graphene, a two-dimensional carbon, has received a rapidly growing research interest. Graphene is the basic building block of carbonaceous materials (3-D graphite, 1-D nanotubes, 0-D fullerenes) and exhibits unique properties comparable with or even better than CNTs. In particular, the advantages of easy preparation by simple chemical or electrochemical processing of graphite,138–141 low resistivity,142 high mechanical strength143 and good SSA144 hold great promise for potential applications of single-layer or few-layer graphene, referred to here as graphene nanosheets (GNS), as supporting material to improve the electrochemical performance of CPs and/or metal oxides.Graphite oxide (GO), treated as important precursor of GNS, can be easily dispersed in water due to a great many hydroxyl, carboxyl and epoxy groups on its basal planes or edges. The negatively charged oxygen-containing groups can be used as nucleation centers to adsorb precursors of CPs or metal oxides onto the GO sheets via electrostatic force. It is believed that good mechanical flexibility of GO can prevent the agglomeration of active materials to even larger particles. On the other hand, the in situ produced NPs adsorbed on the surfaces of exfoliated GO can be used as spacers to readily prevent the close restacking of GO. As a result, the NPs uniformly distribute on the surfaces of GO, which increases both the dispersion of active materials and the contact of active materials with electrolyte, and thus may be favorable for the improvement of the electrochemical performance of an electrode for ECs (Fig. 11).
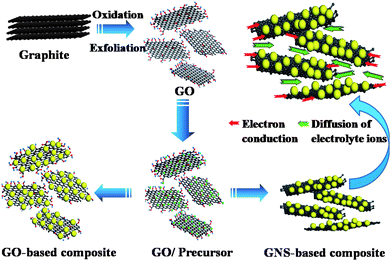 | ||
| Fig. 11 Schematic illustration for the synthesis and the structural advantages of GO or/and GNS-based composites. | ||
Wang et al.145 fabricated a composite of GO supported by needle-like MnO2 nanocrystals (MnO2/GO nanocomposites) through a simple soft chemical route in a water–isopropyl alcohol system. The formation mechanism of the composite was proposed as the intercalation and adsorption of manganese ions onto the GO sheets, then the nucleation and growth of the crystal species in a double solvent system and oriented attachment mechanisms. Hao et al.146 synthesized fibrillar PANI doped with GO via in situpolymerization of monomer in the presence of GO. Although the utilization of MnO2 and PANI in GO-based composites is enhanced, the low conductivity of GO limits further improvement of the active materials. Zhao et al.28 reported that the GO in PANI/GO composites could be reduced to GNS using hydrazine. It was found that the PANI fibers were absorbed on the GNS surfaces or filled between the GNS, and an SC of as high as 480 F g−1 at a current density of 0.1 A g−1 was achieved by GO-doped PANI composites. In this case, post-synthetic treatment is needed to reoxidate and reprotonate of the reduced PANI in order to recover the conductive of PANI. Niu et al.147 reported one-step synthesis of SnO2/GNS composite. They used SnCl2 as reducing agent to reduce graphite oxide (GO) in the presence of HCl and urea. Meanwhile the resultant SnO2 can block the agglomeration of GNS. The electrochemical results exhibit a good supercapacitor performance, whose CV curves were nearly rectangular shape. The SC is 43.4 F g−1 at a scan rate of 0.1 V s−1, moreover, it remains 34.6 F g−1 at a scan rate of 1.0 V s−1 for the SnO2/GNS composite.
In situ synthesis of GNS composites in the presence of GNS is another way to obtain active materials/GNS composites. The GNS in the composites can not only provide highly conductive path, but also serve as a high surface area support material for the deposition of nanometre-sized particles and a tensile backbone to maintain the mechanical strength of the composites. The small nanometre-sized particles can exhibit enhanced electrode/electrolyte interface areas, providing high electroactive regions and short diffusion length, which can ensure the high utilization of active materials (Fig. 11). Manthiram et al.29 found that an SC of 408 F g−1 was obtained for the PANI/GNS composite with 50 wt% PANI loading. Fan et al.148 reported a maximum SC of 1046 F g−1 for the PANI/GNS composite containing 15 wt% GNS at a scan rate of 1 mV s−1. Compared with the GO-guided synthesis, this GNS-guided synthesis is simple, because the cumbersome procedure of reoxidation and reprotonation of the reduced PANI is not required. Fan et al.149 also synthesized the GNS/CNTs/PANI composite, in which the GNS were used as support materials for deposition of PANI particles and CNTs as conductive wires interconnected among GNS/PANI particles. Though a small amount of CNTs (1 wt%) was added into GNS, the cycle stability of GNS/CNTs/PANI composite was greatly improved due to the maintenance of highly conductive path as well as mechanical strength of the electrode during doping-dedoping processes. After 1000 cycles, the capacitance decreased only 6% of the initial capacitance compared to 52% and 67% for GNS/PANI and CNTs/PANI composites, respectively. Dai et al.150 showed a two-step method for growing Ni(OH)2 nanocrystals on GNS with various degrees of oxidation. The single-crystalline Ni(OH)2 hexagonal nanoplates directly grew on GNS and exhibited a high SC of ca. 1335 F g−1 at a charge–discharge current density of 2.8 A g−1 and even ca. 953 F g−1 at 45.7 A g−1 with excellent cycling ability. The high SC and remarkable rate capability are promising for applications in ECs with both high energy and power densities. They pointed out that the direct growth of nanomaterials on GNS is important, which can impart intimate interactions and efficient charge transport between the active nanomaterials and the conducting GNS network. Also, the electrochemical performance of these composites is dependent on the quality of GNS substrates and the morphology and crystallinity of the nanomaterials grown on the surfaces of GNS.
With the growing demand for portable systems, the flexible carbon-based composites have important potential applications as the flexible electrodes in energy storage devices due to their attractive electrochemical properties.130,131 GNS-based sheets are very flexible in contrast to many other paperlike materials, and can be processed easily into film materials.151–153 Accordingly, it is believed that GNS are appropriate candidates as nanoscale substrates to obtain composite electrode films. Cheng et al.154 have prepared freestanding and flexible GNS/PANI composite paper by an in situ anodic electropolymerization of PANI film on GNS paper. This GNS-based composite paper electrode shows a favorable tensile strength of 12.6 MPa and a stable and large electrochemical capacitance (233 F g−1 and 135 F cm−3 for gravimetric and volumetric capacitances), which outperforms many other currently available carbon-based flexible electrodes and is hence particularly promising for flexible ECs. Shi et al.30 reported that composite films of GNS and PANI nanofibers were prepared by vacuum filtration of the mixed dispersions of both components. The composite film has a layered structure, and PANI nanofibers are sandwiched between GNS layers. ECs based on this conductive flexible composite film showed a large electrochemical capacitance (210 F g−1) at a discharge rate of 0.3 A g−1. They also exhibited greatly improved electrochemical stability and rate performance.
The exploration toward the application of GNS-based electrode materials has just begun. Many challenges and opportunities remain. For example, GNS through the chemical reduction of exfoliated GO has contained a significant amount of oxygen and, possibly, significant numbers of defects, which inevitably effects the electrical conductivity of GNS-based electrode materials. Furthermore, new methods are needed to synthesize more conducting GNS with the desired number of layers on a large scale. Additionally, the hierarchical structure of GNS must be hold to supply high SSA for CPs and metal oxides.
4.5 Other carbon forms-based composites
Other carbon structures such as carbon aerogels (CA),155carbon black (CB),156 Ketjen Black (KB),18mesocarbon microbeads (MCMB)157 and carbon fibers,158etc. have also been studied for ECs applications. They possess highly accessible SSA and good electrical conductivity.Carbon aerogels (CA) are considered to be perfect electrode materials for EDLCs due to their unique three-dimensional nano-network, high electrical conductivity, abundant mesopores, controllable pore structure and high SSA.155,159,160 CA-based ECs possess long cycle life and fast response, but usually have lower SC, so there have been many considerable efforts to improve the SCs of CA-based ECs electrodes. Fu et al.161 synthesized nano-sized amorphous α-MnO2 dispersed onto CA via liquid phase co-precipitation technique. The as-prepared CA particles with 20–30 nm in diameter and a narrow PSD of 15 nm were interconnected into a three-dimensional network, while the MnO2 nanomaterials presented filamentous shape. As a result, the synthesized MnO2/CA composite can be considered to be a promising electrode material with the highest SC of 219 F g−1 for ECs. Tong et al.162 explored a novel route to prepare mesoporous MnO2/CA composites using electrochemical deposition assisted by gas bubbles. The obtained composites had a large SSA of 120 m2 g−1, uniform PSD of ca. 5 nm, high SC of 515.5 F g−1, and good stability over 1000 cycles. As we all know, the whole charging/discharging process of the electrode material generally involves: (i) cation transport in the electrolyte; (ii) adsorption/desorption of cations at the surface sites of electrodes, which may be dependent on the ion size and the dehydration/hydration rate; and (iii) cation extraction/insertion into the solid matrix. Whatever the shapes of the active materials, because of the incorporation of CA, the obtained composites always take on a mesoporous structure. On one hand, the three-dimensional nano-network of CA offers abundant connected sites and the active material can be formed and well dispersed, so the effective SSA of electrode is improved sharply and more electroactive sites are exposed. On the other hand, the conductivity of the active material in the composite can be improved. Finally, the ionic/electronic transmission will be more convenient due to the mesoporous structure with 3-D network of the synthesized composite, and as a result, the electrochemical performance could be greatly improved.
Carbon black (CB) is another support for depositing electrode materials to enhance the material utilization. Kim and co-workers used a microemulsion process to prepare MnO2/CB composite.163 The MnO2 in the composite showed a nanorod shape of 2 nm × 10 nm in size. The as-prepared composite showed an SC of 165 F g−1 and gave highly stable and reversible performance for up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Stevanović et al.19 prepared hydrous RuO2/CB nanocomposites via a sol–gel process. The obtained composites prepared from small oxide particles and low-surface-area carbon blacks demonstrated the best energy storage performances at low charging/discharging rates. Like most of carbon materials, CB possesses a surface for active material to disperse and/or grow, provides a low resistance path within the electrode and the pores of CB can increase the ionic conductivity. Sometimes, owing to the existence of CB, the active material is limited to further growth. Because of this, the composite exhibits better electrochemical performance.
000 cycles. Stevanović et al.19 prepared hydrous RuO2/CB nanocomposites via a sol–gel process. The obtained composites prepared from small oxide particles and low-surface-area carbon blacks demonstrated the best energy storage performances at low charging/discharging rates. Like most of carbon materials, CB possesses a surface for active material to disperse and/or grow, provides a low resistance path within the electrode and the pores of CB can increase the ionic conductivity. Sometimes, owing to the existence of CB, the active material is limited to further growth. Because of this, the composite exhibits better electrochemical performance.
Ketjen Black (KB) with a high SSA of 1480 m2 g−1 could be used to combine with metal oxide to improve the rate performance of the electrode, such as RuO2.164 Kim et al.165 prepared nanostructured amorphous RuO2·xH2O/KB composite with 40 wt% of RuO2 contents via a modified sol–gel method using glycolic acid as a stabilizer. The composite with RuO2 NPs of ca. 2 nm showed an SC of 462 F g−1. Naoi et al.18 prepared nanosized RuO2/KB composites using an in situ sol–gel process induced by ultracentrifugal (UC) mechanical force, which had a high SC of 821 F g−1 due to the high dispersion of RuO2 into KB. A model capacitor assembled using the composite exhibited energy and power densities as high as 12 Wh kg−1 and 6 kW kg−1, respectively. Fig. 12 schematically shows the possible mechanism for producing the composite with unique structure by a simple UC process. First, the precursors and KB are mixed by ultrasonic wave, where the precursor is supposed to attach on the hollow carbon (KB) surfaces. Secondly, products are formed by addition of alkali, then graphene layers of the KB are milled and partially broken up under UC force for several minutes. Thirdly, the fractured hollow graphenes reaggregate along with encapsulation of the the product particles when the shear of the UC stress is discontinued. The composites with this kind of structure can promote the utilization of active material due to their high surface area with much active sites inside. Therefore, KB is thought to be a promising carbon substrate for ECs applications.
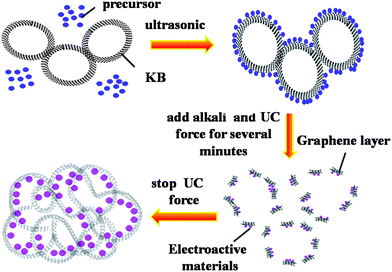 | ||
| Fig. 12 Possible mechanism for electroactive NPs encapsulated in KB by the UC process. | ||
Because mesocarbon microbeads (MCMB) have a unique spherical structure with a diameter of approximately 1–40 μm, the activating products (AMCMB), generally are unique fine granules with deficient sphericity, which have potential applications in ECs.166MCMB treated by a hydrothermal process as a support for NiO were first reported by Zhang et al.,157 the NiO/h-MCMB (hydrothermal treated mesocarbon microbeads) composites with 23 wt% NiO loading could achieve an SC of ca. 637 F g−1. Li et al.167 reported activated Mn3O4/MCMB showing a maximum SC of 178 F g−1 at a current density of 330 mA g−1.
Besides, there are still some other carbon supports. Wen et al.168 prepared polyaniline-implanted porous carbon electrode material by electropolymerization exhibiting an SC of as high as 160 F g−1. Ma et al.169 synthesized manganese oxide/acetylene black via spontaneously deposition for application in ECs.
5. Summary and future outlook
In this review, we attempted to highlight the latest researches of constructive design, interesting synthesis and good electrochemical performance of carbon-based composites with hierarchical structures for ECs applications, from which some directive strategies can be deduced to guide the synthesis of electroactive materials with good electrochemical capacitance. Herein, the hierarchically structured carbon-based composites represent very exciting materials for ECs as they constitute a complex and unique architecture that provides a large SSA, excellent conductance as well as fast ion transportation for sufficient electrochemical reactions at their interface. The major direction for future research in this area lies in elaborately designing, controlling and tailoring an exciting and even simpler strategy to synthesize novel carbon-based composites in hierarchical levels, and to realize and exploit their more desirable functions for ECs application.One of the great challenges in the development of ECs technology is their relatively high cost when compared to other energy devices. The cost of these novel hierarchical composites will be greatly cut down with the rapid development of nanotechnology, which is of great importance in their practical application. Of note, electrolytes also play a great role in the enhancement of the overall electrochemical performance of ECs. In our laboratory,170 the Fe(CN)63−/Fe(CN)64− ion pair was originally introduced into the alkaline KOH electrolyte to enhance the energy storage of the Co–Al layered double hydroxide (Co–Al LDH) electrode, which is also versatile in other systems. Therefore, we also expect the proper design of electrolytes for further optimizing electrochemical behavior of hierarchically structured carbon-based composites, finally resulting in excellent electrochemical capacitance for an EC. Moreover, the detailed discussion on the relationship between the specific structures (pore structure, SSA, electronic conductivity, etc.) of the as-designed electroactive materials with hierarchical structures and the electrochemical performance of the as-fabricated ECs based on these electrodes, to the best of our knowledge, is scarcely reported, however the relationship is also important for ECs’ applications, and a vast field is left to be exploited. Wang et al.171 constructed asymmetric capacitors composed of carbon electrodes with different PSD in order to study the fundamental relationship between the PSD of positive and negative carbon electrodes and the performance of EDLCs using a (C2H5)3CH3NBF4/propylenecarbonateelectrolyte solution. The negative electrode carbon of the asymmetric EDLCs governed its capacitive characteristics and the presence of mesopores in the negative electrode carbon was important for the whole performance of asymmetric EDLCs with non-aqueous electrolyte, particularly for high-rate performance.
All in all, there is tremendous room still left in the design and synthesis of hierarchically structured carbon-based composites with good electrochemical capacitance to be explored by the chemists, electrochemists and theoreticians and it is believed that any breakthroughs in the area will spark the development of promising ECs with a larger energy density and good electrochemical stability at high rates with an in-depth understanding of their specific structure and dynamic information at the electrode interface in the near future.
Acknowledgements
This work was supported by National Basic Research Program of China (973 Program) (No. 2007CB209703), National Natural Science Foundation of China (No.206033040, No.20873064).References
- J. Tollefson, Nature, 2008, 456, 436–440 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic Publishers/Plenum Press, New York, 1999, ch. 20 Search PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS.
- J. W. Long, Electrochem. Soc. Interface Spring, 2008, 17, 33 Search PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. R. Miller and A. F. Burke, Electrochem. Soc. Interface Spring, 2008, 17, 53–57 Search PubMed.
- H. Zhang, G. P. Cao and Y. S. Yang, Energy Environ. Sci., 2009, 2, 932–943 RSC.
- C. Z. Yuan, S. L. Xiong, X. G. Zhang, L. F. Shen, F. Zhang, B. Gao and L. H. Su, Nano Res., 2009, 2, 722–732 CrossRef CAS.
- H. L. Wang, J. T. Robinson, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS.
- J. Liu, G. Z. Cao, Z. G. Yang, D. H. Wang, D. Dubois, X. D. Zhou, G. L. Graff, L. R. Pederson and J. G. Zhang, ChemSusChem, 2008, 1, 676–697 CrossRef CAS.
- Y. G. Wang, H. Q. Li and Y. Y. Xia, Adv. Mater., 2006, 18, 2619–2623 CrossRef CAS.
- X. Du, C. Y. Wang, M. M. Chen, Y. Jiao and J. Wang, J. Phys. Chem. C, 2009, 113, 2643–2646 CrossRef CAS.
- H. Zhang, G. P. Cao, Z. Y. Wang, Y. S. Yang, Z. J. Shi and Z. N. Gu, Nano Lett., 2008, 8, 2664–2668 CrossRef CAS.
- S. Mitra, K. S. Lokesh and S. Sampath, J. Power Sources, 2008, 185, 1544–1549 CrossRef CAS.
- K. Naoi, S. Ishimoto, N. Ogihara, Y. Nakagawa and S. Hatta, J. Electrochem. Soc., 2009, 156, A52–A59 CrossRef CAS.
- V. V. Panic, A. B. Dekanski and R. M. Stevanovic, J. Power Sources, 2010, 195, 3969–3976 CrossRef CAS.
- C. Z. Yuan, L. Chen, B. Gao, L. H. Su and X. G. Zhang, J. Mater. Chem., 2009, 19, 246–252 RSC.
- C. Masarapu, H. F. Zeng, K. H. Hung and B. Q. Wei, ACS Nano, 2009, 3, 2199–2206 CrossRef CAS.
- Y. Y. Liang, X. L. Feng, L. J. Zhi, U. Kolb and K. Müllen, Chem. Commun., 2009, 809–811 RSC.
- B. Xu, F. Wu, R. Chen, G. Cao, S. Chen, Z. Zhou and Y. Yang, Electrochem. Commun., 2008, 10, 795–797 CrossRef CAS.
- C. Kim, B. T. N. Ngoc, K. S. Yang, M. Kojima, Y. A. Kim, Y. J. Kim, M. Endo and S. C. Yang, Adv. Mater., 2007, 19, 2341–2346 CrossRef CAS.
- C. Z. Yuan, B. Gao and X. G. Zhang, J. Power Sources, 2007, 173, 606–612 CrossRef CAS.
- E. Raymundo-pinero, F. Leroux and F. Beguin, Adv. Mater., 2006, 18, 1877–1882 CrossRef CAS.
- M. Zhu, C. J. Weber, Y. Yang, M. Konuma, U. Starke, K. Kern and A. M. Bittner, Carbon, 2008, 46, 1829–1840 CrossRef CAS.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. S. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 21, 5004–5006 CrossRef CAS.
- Q. Wu, Y. X. Xu, Z. Y. Yao, A. R Liu and G. Q. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS.
- C. O. Ania, V. Khomenko, E. Raymundo-Pinero, J. B. Parra and F. Beguin, Adv. Funct. Mater., 2007, 17, 1828–1836 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373–376 CrossRef CAS.
- X. P. Dong, W. H. Shen, J. L. Gu, L. M. Xiong, Y. F. Zhu, H. Li and J. L. Shi, J. Phys. Chem. B, 2006, 110, 6015–6019 CrossRef CAS.
- C. C. Hu, K. H. Chang, M. C. Lin and Y. T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS.
- C. Z. Yuan, L. H. Su, B. Gao and X. G. Zhang, Electrochim. Acta, 2008, 53, 7039–7047 CrossRef CAS.
- S. L. Xiong, C. Z. Yuan, X. G. Zhang, B. J. Xi and Y. T. Qian, Chem.–Eur. J., 2009, 15, 5320–5326 CrossRef CAS.
- C. Z. Yuan, X. G. Zhang, L. H. Su, B. Gao and L. F. Shen, J. Mater. Chem., 2009, 19, 5772–5777 RSC.
- D. Choi, G. E. Blomgren and P. N. Kumta, Adv. Mater., 2006, 18, 1178–1182 CrossRef CAS.
- C. Z. Yuan, B. Gao, L. H. Su, L. Chen and X. G. Zhang, J. Electrochem. Soc., 2009, 156, A199–A203 CrossRef CAS.
- H. Y. Mi, X. G. Zhang, S. Y. An, X. G. Ye and S. D. Yang, Electrochem. Commun., 2007, 9, 2859–2862 CrossRef CAS.
- B. Gao, Q. B. Fu, L. H. Su, C. Z. Yuan and X. G. Zhang, Electrochim. Acta, 2010, 55, 2311–2318 CrossRef CAS.
- H. Y. Mi, X. G. Zhang, X. G. Ye and S. D. Yang, J. Power Sources, 2008, 176, 403–409 CrossRef CAS.
- P. Soudan, P. Lucas, H. A. Ho, D. Jobin, L. Breau and D. Belanger, J. Mater. Chem., 2001, 11, 773–782 RSC.
- L. Chen, C. Z. Yuan, H. Dou, B. Gao, S. Y. Chen and X. G. Zhang, Electrochim. Acta, 2009, 54, 2335–2341 CrossRef CAS.
- W. Dong, D. R. Rolison and B. Dunn, Electrochem. Solid-State Lett., 2000, 3, 457–459 CrossRef CAS.
- H. V. Helmholtz, Ann. Phys., 1853, 89, 211–233 Search PubMed.
- G. Gouy, J. Phys., 1910, 4, 457–468.
- D. L. Chapman, Philos. Mag., 1913, 6, 475–481.
- C. W. Huang, Y. T. Wu, C. C. Hu and Y. Y. Li, J. Power Sources, 2007, 172, 460–467 CrossRef CAS.
- H. S. Zhou, S. M. Zhu, M. Hibino and I. Honma, J. Power Sources, 2003, 122, 219–223 CrossRef CAS.
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowski and F. Beguin, Carbon, 2006, 44, 2498–2507 CrossRef CAS.
- J. Huang, B. G. Sumpter and V. Meunier, Chem.–Eur. J., 2008, 14, 6614–6626 CrossRef CAS.
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539–1548 CAS.
- B. E. Conway and D. Craig, Report to Continental Group Inc. No. 1, 1975 Search PubMed, ref. 5.
- B. E. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef CAS.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- K. Kierzek, E. Frackowiak, G. Lota, G. Gryglewicz and J. Machnikowski, Electrochim. Acta, 2004, 49, 1169–1170 CrossRef CAS.
- E. Raymundo-Pinero, F. Leroux and F. Beguin, Adv. Mater., 2006, 18, 1877–1882 CrossRef CAS.
- J. R. Zhang, D. C. Jiang, B. Chen, J. J. Zhu, L. P. Jiang and H. Q. Fang, J. Electrochem. Soc., 2001, 148, A1362–A1367 CrossRef CAS.
- T. Nanaumi, Y. Ohsawa, K. Kobayakawa and Y. Sato, Electrochemistry, 2002, 70, 681–685 CAS.
- M. S. Dandekar, G. Arabale and K. Vijayamohanan, J. Power Sources, 2005, 141, 198–203 CrossRef CAS.
- Y. F. Su, F. Wu, L. Y. Bao and Z. H. Yang, New Carbon Mater., 2007, 22, 53–57 Search PubMed.
- G. H. Yuan, Z. H. Jiang, A. Aramata and Y. Z. Gao, Carbon, 2005, 43, 2913–2917 CrossRef CAS.
- T. Morishita, Y. Soneda, T. Tsumura and M. Inagaki, Carbon, 2006, 44, 2360–2367 CrossRef CAS.
- V. Ganesh, S. Pitchumani and V. Lakshminarayanan, J. Power Sources, 2006, 158, 1523–1532 CrossRef CAS.
- Q. H. Huang, X. Y. Wang, J. Li, C. L. Dai, S. Gamboa and P. J. Sebastian, J. Power Sources, 2007, 164, 425–429 CrossRef CAS.
- K. Okajima, A. Ikeda, K. Kamoshita and M. Sudoh, Electrochim. Acta, 2005, 51, 972–977 CrossRef CAS.
- H. Q. Li, Y. Zou and Y. Y. Xia, Electrochim. Acta, 2007, 52, 2153–2157 CrossRef CAS.
- H. Tamai, M. Hakoda, T. Shiono and H. Yasuda, J. Mater. Sci., 2007, 42, 1293–1298 CrossRef CAS.
- Q. Wang, J. L. Li, F. Gao, W. S. Li, K. Z. Wu and X. D. Wang, New Carbon Mater., 2008, 23, 275–280 Search PubMed.
- D. Y. Qu and H. Shi, J. Power Sources, 1998, 74, 99–107 CrossRef CAS.
- C. L. Liang, Z. J. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS.
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. B. Kim, Chem. Commun., 1999, 2177–2178 RSC.
- H. S. Zhou, S. M. Zhu, M. Hibino and I. Honma, J. Power Sources, 2003, 122, 219–223 CrossRef CAS.
- S. Alvarez, M. C. Blanco-Lopez, A. J. Miranda-Ordieres, A. B. Fuertes and T. A. Centeno, Carbon, 2005, 43, 866–870 CrossRef CAS.
- W. Xing, S. Z. Qiao, R. G. Ding, F. Li, G. Q. Lu, Z. F. Yan and H. M. Cheng, Carbon, 2006, 44, 216–224 CrossRef CAS.
- H. Yamada, H. Nakamura, F. Nakahara, I. Moriguchi and T. Kudo, J. Phys. Chem. C, 2007, 111, 227–233 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS.
- R. Lin, P. L. Taberna, J. Chmiola, D. Guay, Y. Gogotsi and P. Simon, J. Electrochem. Soc., 2009, 156, A7–A12 CrossRef CAS.
- W. Xing, S. P Zhuo, H. Y. Cui and Z. F. Yan, Mater. Lett., 2007, 61, 4627–4630 CrossRef CAS.
- L. X. Li, H. H. Song, Q. C. Zhang, J. Y. Yao and X. H. Chen, J. Power Sources, 2009, 187, 268–274 CrossRef CAS.
- J. Zhang, L. B. Kong, J. J. Cai, H. Li, Y. C. Luo and L. Kang, Microporous Mesoporous Mater., 2010, 132, 154–162 CrossRef CAS.
- Y. L. Cao, J. M. Cao, M. B. Zheng, J. S. Liu and G. B. Ji, J. Solid State Chem., 2007, 180, 792–798 CrossRef CAS.
- X. P. Dong, W. H. Shen, J. L. Gu, L. M. Xiong, Y. F. Zhu, Z. Li and J. L. Shi, J. Phys. Chem. B, 2006, 110, 6015–6019 CrossRef CAS.
- H. F. Li, R. D. Wang and R. Cao, Microporous Mesoporous Mater., 2008, 111, 32–38 CrossRef CAS.
- F. Pico, E. Morales, J. A. Fernandez, T. A. Centeno, J. Ibanez, R. M. Rojas, J. M. Amarilla and J. M. Rojo, Electrochim. Acta, 2009, 54, 2239–2245 CrossRef CAS.
- B. T. Holland, C. F. Blanford and A. Stein, Science, 1998, 281, 538–540 CrossRef CAS.
- S. W. Woo, K. Dokko, H. Nakano and K. Kanamura, J. Power Sources, 2009, 190, 596–600 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus and P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic, New York/San Diego, 1996 Search PubMed.
- M. S. Dresselhaus, G. Dresselhaus and R. Saito, Solid State Commun., 1992, 84, 201–204 CrossRef CAS.
- J. P. Issi, L. Langer, J. Heremans and C. H. Olk, Carbon, 1995, 33, 941–948 CrossRef CAS.
- T. W. Ebbesen, H. J. Lezec, H. Hiura, J. W. Bennett, H. F. Ghaemi and T. Thio, Nature, 1996, 382, 54–56 CrossRef CAS.
- C. Niu, E. K. Sichel, R. Hoch, D. Moy and H. Tennent, Appl. Phys. Lett., 1997, 70, 1480–1482 CrossRef CAS.
- K. H. An, W. S. Kim, Y. S. Park, Y. C. Choi, S. M. Lee, D. C. Chung, D. J. Bae and S. C. Lim, Adv. Mater., 2001, 13, 497–500 CrossRef CAS.
- C. S. Du and N. Pan, Nanotechnology, 2006, 17, 5314–5318 CrossRef CAS.
- E. Frackowiak, K. Metenier, V. Bertagna and F. Beguin, Appl. Phys. Lett., 2000, 77, 2421–2423 CrossRef CAS.
- A. L. M. Reddy, F. E. Amitha, I. Jafri and S. Ramaprabhu, Nanoscale Res. Lett., 2008, 3, 145–151 CrossRef.
- C. G. Liu, M. Liu, F. Li and H. M. Cheng, Appl. Phys. Lett., 2008, 92, 143108 CrossRef.
- J. N. Barisci, G. G. Wallace, D. Chattopadhyay, F. Papadimitrakopoulos and R. H. Baughman, J. Electrochem. Soc., 2003, 150, E409–E415 CrossRef CAS.
- Y. T. Kim, Y. Ito, K. Tadai, T. Mitani, U. S. Kim, H. S. Kim and B. W. Cho, Appl. Phys. Lett., 2005, 87, 234106 CrossRef.
- E. Frackowiak, S. Delpeux, K. Jurewicz, K. Szostak, D. Cazorla-Amoros and F. Beguin, Chem. Phys. Lett., 2002, 361, 35–41 CrossRef CAS.
- Y. J. Kim, Y. A. Kim, T. Chino, T. Chino, H. Suezaki, M. Endo and M. S. Dresselhaus, Small, 2006, 2, 339–345 CrossRef CAS.
- Y. T. Kim and T. Mitani, J. Power Sources, 2006, 158, 1517–1522 CrossRef CAS.
- B. Xu, F. Wu, Y. F. Su, G. P. Cao, C. Shi, Z. M. Zhou and Y. S. Yang, Electrochim. Acta, 2008, 53, 7730–7735 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- L. H. Su, X. G. Zhang, C. Z. Yuan and B. Gao, J. Electrochem. Soc., 2008, 155, A110–A114 CrossRef CAS.
- C. Y. Lee, H. M. Tsai, H. J. Chuang, S. Y. Li, P. Lin and T. Y. Tseng, J. Electrochem. Soc., 2005, 152, A716–A720 CrossRef CAS.
- G. Arabale, D. Wagh, M. Kulkarni, I. S. Mulla, S. P. Vernekar, K. Vijayamohanan and A. M. Rao, Chem. Phys. Lett., 2003, 376, 207–213 CrossRef CAS.
- K. Liang, K. An and Y. Lee, J. Mater. Sci. Tech., 2005, 21, 292–296 CAS.
- M. Hughes, G. Z. Chen, M. S. P. Shaffer, D. J. Fray and A. H. Windle, Chem. Mater., 2002, 14, 1610–1613 CrossRef CAS.
- J. H. Park, J. M. Ko and O. O. Park, J. Electrochem. Soc., 2003, 150, A864–A867 CrossRef CAS.
- Y. T. Kim, K. Tadai and T. Mitani, J. Mater. Chem., 2005, 15, 4914–4921 RSC.
- Y. T. Kim and T. Mitani, Appl. Phys. Lett., 2006, 89, 033107 CrossRef.
- W. C. Fang, M. S. Leu, K. H. Chen and L. C. Chen, J. Electrochem. Soc., 2008, 155, K15–K18 CrossRef CAS.
- W. C. Fang, K. H. Chen and L. C. Chen, Nanotechnology, 2007, 18, 485716 CrossRef.
- Y. G. Wang, L. Yu and Y. Y. Xia, J. Electrochem. Soc., 2006, 153, A743–A748 CrossRef CAS.
- B. Gao, L. Hao, Q. B. Fu, L. H. Su, C. Z. Yuan and X. G. Zhang, Electrochim. Acta, 2010, 55, 3681–3686 CrossRef CAS.
- B. Gao, C. Z. Yuan, L. H. Su, L. Chen and X. G. Zhang, J. Solid State Electrochem., 2009, 13, 1251–1257 CrossRef CAS.
- M. Baibarac, I. Baltog, C. Godon, S. Lefrant and O. Chauvet, Carbon, 2004, 42, 3143–3152 CrossRef CAS.
- T. M. Wu, Y. W. Lin and C. S. Liao, Carbon, 2005, 43, 734–740 CrossRef CAS.
- E. Frackowiak, V. Khomenkob, K. Jurewicza, K. Lota and F. Beguin, J. Power Sources, 2006, 153, 413–418 CrossRef CAS.
- V. Gupta and N. Miura, Electrochim. Acta, 2006, 52, 1721–1726 CrossRef CAS.
- J. E. Huang, X. H. Li, J. C. Xu and H. L. Li, Carbon, 2003, 41, 2731–2736 CrossRef CAS.
- R. Bandyopadhyaya, E. Nativ-Roth, O. Regev and R. Yerushalmi-Rozen, Nano Lett., 2002, 2, 25–28 CrossRef CAS.
- R. K. Sharma and L. Zhai, Electrochim. Acta, 2009, 54, 7148–7155 CrossRef CAS.
- R. K. Sharma, A. Karakoti, S. Seal and L. Zhai, J. Power Sources, 2010, 195, 1256–1262 CrossRef CAS.
- L. B. Hu, H. Wu and Y. Cui, Appl. Phys. Lett., 2010, 96, 183502 CrossRef.
- J. Chen, A. I. Minett, Y. Liu, C. Lynam, P. Sherrell, C. Y. Wang and G. G. Wallace, Adv. Mater., 2008, 20, 566–570 CrossRef CAS.
- S. L. Chou, J. Z. Wang, S. Y. Chew, H. K. Liu and S. X. Dou, Electrochem. Commun., 2008, 10, 1724–1727 CrossRef CAS.
- Y. Y. Horng, Y. C. Lu, Y. K. Hsu, C. C. Chen, L. C. Chen and K. H. Chen, J. Power Sources, 2010, 195, 4418–4422 CrossRef CAS.
- H. Zhang, G. P. Cao, Y. S. Yang and Z. N. Gu, J. Electrochem. Soc., 2008, 155, K19–K22 CrossRef CAS.
- L. J. Gao, A. P. Peng, Z. Y. Wang, H. Zhang, Z. J. Shi, Z. N. Gu, G. P. Cao and B. Z. Ding, Solid State Commun., 2008, 146, 380–383 CrossRef CAS.
- C. S. Du and N. Pan, J. Power Sources, 2006, 160, 1487–1497 CrossRef CAS.
- C. S. Du, J. Yeh and N. Pan, Nanotechnology, 2005, 16, 350–353 CrossRef CAS.
- H. Zhang, G. P. Cao and Y. S. Yang, Nanotechnology, 2007, 18, 195607 CrossRef.
- H. Zhang, G. P. Cao, Z. Y. Wang, Y. S. Yang, Z. J. Shi and Z. N. Gu, Electrochem. Commun., 2008, 10, 1056–1059 CrossRef CAS.
- N. Liu, F. Luo, H. X. Wu, Y. H. Liu, C. Zhang and J. Chen, Adv. Funct. Mater., 2008, 18, 1518–1525 CrossRef CAS.
- G. X. Wang, B. Wang, J. Park, Y. Wang, B. Sun and J. Yao, Carbon, 2009, 47, 3242–3246 CrossRef CAS.
- G. H. Chen, W. G. Weng, D. J. Wu, C. L. Wu, J. R. Lu, P. P. Wang and X. F. Chen, Carbon, 2004, 42, 753–759 CrossRef CAS.
- L. M. Viculis, J. J. Mack, O. M. Mayer, H. T. Hahn and R. B. Kaner, J. Mater. Chem., 2005, 15, 974–978 RSC.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. Okeeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS.
- S. Chen, J. W. Zhu, X. D. Wu, Q. F. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS.
- H. L. Wang, Q. L. Hao, X. J. Yang, L. D. Lu and X. Wang, Electrochem. Commun., 2009, 11, 1158–1161 CrossRef CAS.
- F. H. Li, J. F. Song, H. F. Yang, S. Y. Gan, Q. X. Zhang, D. X. Han, A. Ivaska and L. Niu, Nanotechnology, 2009, 20, 455602 CrossRef.
- J. Yan, T. Wei, B. Shao, Z. J. Fan, W. Z. Qian, M. L. Zhang and F. Wei, Carbon, 2010, 48, 487–493 CrossRef CAS.
- J. Yan, T. Wei, Z. J. Fan, W. Z. Qian, M. L. Zhang, X. D. Shen and F. Wei, J. Power Sources, 2010, 195, 3041–3045 CrossRef CAS.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS.
- S. Park, K. S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS.
- H. Q. Chen, M. B. Muller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS.
- D. W. Wang, F. Li, J. P. Zhao, W. C. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745–1752 CrossRef CAS.
- S. T. Mayer, R. W. Pekala and J. L. Kaschmitter, J. Electrochem. Soc., 1993, 140, 446–451 CAS.
- S. L. Kuo and N. L. Wu, J. Power Sources, 2006, 162, 1437–1443 CrossRef CAS.
- C. Z. Yuan, B. Gao, L. H. Su and X. G. Zhang, Solid State Ionics, 2008, 178, 1859–1866 CrossRef CAS.
- E. J. Ra, E. Raymundo-Pinero, Y. H. Lee and F. Beguin, Carbon, 2009, 47, 2984–2992 CrossRef CAS.
- D. C. Wu, R. W. Fu, M. S. Dresselhaus and G. Dresselhaus, Carbon, 2006, 44, 675–681 CrossRef CAS.
- J. Li, X. Y. Wang, Q. H. Huang, S. Gamboa and P. J. Sebastian, J. Power Sources, 2006, 158, 784–788 CrossRef CAS.
- G. F. Lv, D. C. Wu and R. W. Fu, J. Non-Cryst. Solids, 2009, 355, 2461–2465 CrossRef CAS.
- G. R. Li, Z. P. Feng, Y. N. Ou, D. C. Wu, R. W. Fu and Y. X. Tong, Langmuir, 2010, 26, 2209–2213 CrossRef CAS.
- R. K. Sharma, H. S. Oh, Y. G. Shul and H. Kim, J. Power Sources, 2007, 173, 1024–1028 CrossRef CAS.
- M. Min, K. Machida, J. H. Jang and K. Naoi, J. Electrochem. Soc., 2006, 153, A334–A338 CrossRef CAS.
- Y. H. Lee, J. G. Oh, H. S. Oh and H. Kim, Electrochem. Commun., 2008, 10, 1035–1037 CrossRef CAS.
- Z. M. Shen and R. S. Xue, Fuel Process. Technol., 2003, 84, 95–103 CrossRef CAS.
- H. Q. Wang, Z. S. Li, J. H. Yang, Q. Y. Li and X. X. Zhong, J. Power Sources, 2009, 194, 1218–1221 CrossRef CAS.
- W. C. Chen and T. C. Wen, J. Power Sources, 2003, 117, 273–282 CrossRef CAS.
- S. B. Ma, Y. H. Lee, K. Y. Ahn, C. M. Kim, K. H. Oh and K. B. Kim, J. Electrochem. Soc., 2006, 153, C27–C32 CrossRef CAS.
- L. H. Su, X. G. Zhang, C. H. Mi, B. Gao and Y. Liu, Phys. Chem. Chem. Phys., 2009, 11, 2195–2202 RSC.
- L. H. Wang, T. Morishita, M. Toyoda and M. Inagaki, Electrochim. Acta, 2007, 53, 882–886 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |