Abiotic formation of uroporphyrinogen and coproporphyrinogen from acyclic reactants†
Jonathan S.
Lindsey
*,
Vanampally
Chandrashaker
,
Masahiko
Taniguchi
and
Marcin
Ptaszek
Department of Chemistry, North Carolina State University, Raleigh, NC 27695, USA. E-mail: jlindsey@ncsu.edu
First published on 6th December 2010
Abstract
Tetrapyrrole macrocycles (e.g., porphyrins) have long been proposed as key ingredients in the emergence of life, yet plausible routes for forming their essential pyrrole precursor have previously not been identified. Here, the anaerobic reaction of δ-aminolevulinic acid (ALA, 5–240 mM) with 5-methoxy-3-(methoxyacetyl)levulinic acid (1-AcOH, 5–240 mM) in water (pH 5–7) at 25–85 °C for a few hours to a few days affords uroporphyrinogen, which upon chemical oxidation gives uroporphyrin in overall yield of up to 10%. The key intermediate is the α-methoxymethyl-substituted analogue of the pyrrole porphobilinogen (PBG). Reaction of ALA and the decarboxy analogue of 1-AcOH (1-Me) gave coproporphyrinogen (without its biosynthetic precursor uroporphyrinogen as an intermediate); oxidation gave the corresponding coproporphyrin in yields comparable to those for uroporphyrin. In each case a mixture of porphyrin isomers was obtained, consistent with reversible oligopyrromethane formation. The route investigated here differs from the universal extant biosynthetic pathway to tetrapyrrole macrocycles, where uroporphyrinogen (isomer III) – nature's last common precursor to corrins, heme, and chlorophylls – is derived from eight molecules of ALA (via four molecules of PBG). The demonstration of the spontaneous self-organization of eight acyclic molecules to form the porphyrinogen under simple conditions may open the door to the development of a chemical model for the prebiogenesis of tetrapyrrole macrocycles.
Introduction
Tetrapyrrole macrocycles play a central role in most present living organisms yet the prebiotic origin of such molecules remains obscure.1 The dearth of understanding exists despite a recurring view since the 1950s that such molecules were likely to be valuable if not essential for the origin of life.2–18 Such views stem from the unique attributes of tetrapyrroles for diverse bioenergetic processes (including photosynthesis) and have been further encouraged by the universality, concision, and molecular logic of the tetrapyrrole biosynthetic pathway. The early steps of the biosynthesis19 are shown in Fig. 1. Two molecules of ALA dimerize to give porphobilinogen (PBG), which undergoes tetramerization and macrocyclization (accompanied by inversion of the orientation of one pyrrole) to give uroporphyrinogen III. Uroporphyrinogen III is the last common progenitor of the corrinoid, porphyrin, and (bacterio)chlorin macrocycles known as the pigments of life. Decarboxylation of the four acetic acid side chains of uroporphyrinogen III affords coproporphyrinogen III, which upon decarboxylation and dehydrogenation of two of the four propionic acid side chains affords protoporphyrinogen IX. Dehydrogenation (−6e−, −6H+) of the protoporphyrinogen IX macrocycle yields protoporphyrin IX, the ligand for heme and a precursor to all chlorophylls and bacteriochlorophylls.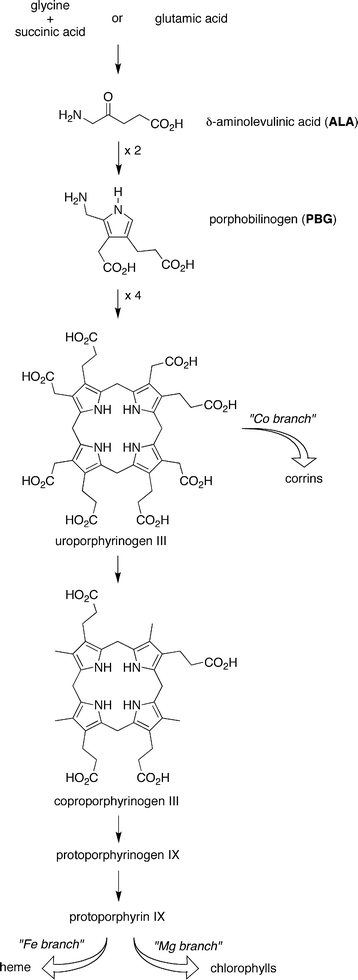 | ||
| Fig. 1 Contemporary biosynthesis of tetrapyrrole macrocycles. | ||
Granick and Mauzerall put forth the view that the biosynthesis provides a “window on evolution” where each molecule served a function in its time and then was replaced with molecules formed later in the biosynthetic pathway as transformations were appended over time.3,4,8,18,20–22 The rationale for this view stems from the multifaceted change in features of the molecules in proceeding from the early to later stages of the biosynthesis: (i) from water-soluble compounds to hydrophobic macrocycles, (ii) from saturated to unsaturated molecules, and (iii) from simple to complex molecular architectures. The evolutionary perspective that “biosynthesis recapitulates biogenesis”20 has prompted consideration as to whether the earliest steps might have been operative in the prebiotic era – via purely structure-directed processes (i.e., without enzymes or other catalysts) – thereby yielding tetrapyrole macrocycles that underpinned early catalysis and proto-photosynthesis.
A proposed prebiotic pathway that closely mirrors the extant biosynthesis is outlined in Fig. 2. Examples of transformations and products in each of the steps include the (1) conversion of early-Earth available substances to ALA, (2) dimerization of ALA to give PBG, (3) tetramerization of PBG and macrocyclization to give uroporphyrinogen (four isomers), (4) dehydrogenation to give uroporphyrin (four isomers), and (5) subsequent metalation, decarboxylation, and/or reduction to form macrocycles such as metalated uroporphyrin, coproporphyrin, and hydroporphyrin (e.g., chlorin, bacteriochlorin) analogues. Uroporphyrin (and uroporphyrinogen) have been proposed as prebiotic photosensitizers given their water solubility, characteristic reactions, and potential for prebiotic formation.9–11,18,22,23 Studies to address the occurrence of the individual steps include the following:
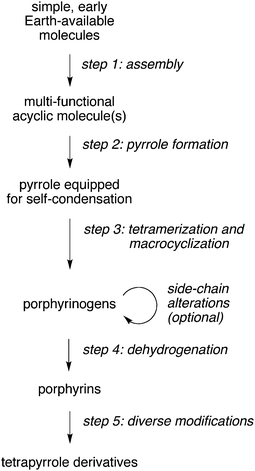 | ||
| Fig. 2 Conceptual steps in a pre-biosynthetic route to tetrapyrroles. | ||
• Diverse modifications (step 5): Uroporphyrin undergoes smooth metalation [e.g., with zinc(II) or copper(II)] in neutral aqueous solution at room temperature;24,25 facile photoreduction to give a variety of porphomethenes;26,27 and, under exceptionally forcing conditions, decarboxylation of the acetic acid side chains.28
• Dehydrogenation (step 4): The dehydrogenation of a porphyrinogen to give the porphyrin can be achieved with oxidants under a variety of conditions.29 The dehydrogenation of uroporphyrinogen can be achieved in aqueous solution by chemical or photochemical oxidation, and moreover, uroporphyrin can catalyze the dehydrogenation in an autocatalytic manner as shown in Fig. 3.30 The autocatalytic process has been demonstrated with oxygen but is expected to proceed with diverse (prebiotic) oxidants.
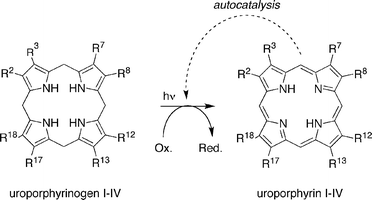 | ||
| Fig. 3 Step 4: photochemical oxidation of uroporphyrinogen affords uroporphyrin in an autocatalytic manner. | ||
• Tetramerization and macrocyclization (step 3): PBG undergoes condensation in the absence of enzymes to give the estimated statistical mixture of four uroporphyrinogen isomers as shown in Fig. 4. Examination of the distribution of uroporphyrinogens (inferred upon oxidation to give the corresponding uroporphyrin isomers) indicates that isomer III is the most abundant, as expected on statistical considerations.31 Although strong acid was predominantly employed for the condensation, a few reactions have been reported under neutral conditions. At pH 7.6 and 60 °C for 21 h under anaerobic conditions, PBG (10 mM) affords the uroporphyrinogen isomers in 70% yield.31 Similarly, PBG (0.26 mM) at pH 7.4 and 70 °C for 2 h under anaerobic conditions affords the uroporphyrinogen isomers in ∼65% yield.32 Treatment of a given uroporphyrinogen isomer to the acidic macrocyclization conditions results in the statistical distribution of isomers, providing evidence for the thermodynamic stability of the macrocycles.33 These data indicate the robustness of the macrocyclization process under plausible prebiotic conditions.
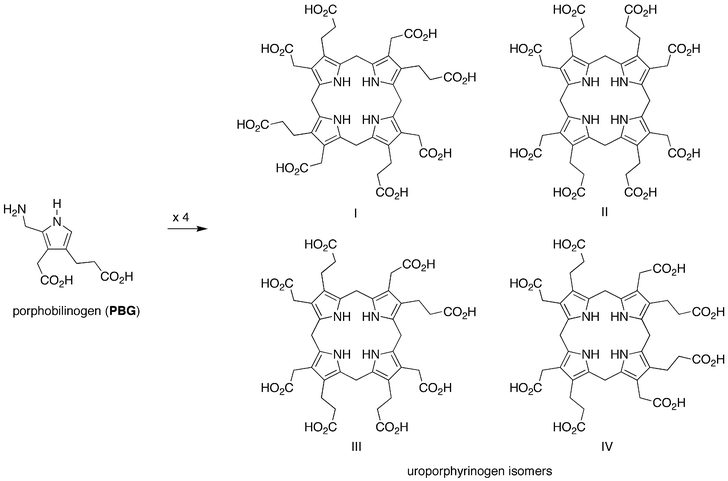 | ||
| Fig. 4 Step 3: solution condensation of porphobilinogen affords four uroporphyrinogen isomers. | ||
• Pyrrole formation (step 2): ALA in solution affords a dihydropyrazine and pseudo-porphobilinogen rather than PBG, the latter outcome owing to the greater reactivity of the δ versus β methylene unit (Fig. 5).34 Despite extensive studies, prebiotically plausible conditions have not been identified for the solution dimerization of ALA to give PBG.35
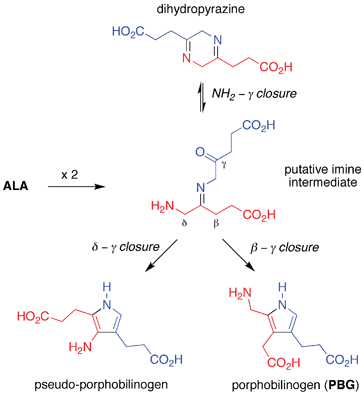 | ||
| Fig. 5 Attempted step 2: solution self-condensation of ALA affords pseudo-porphobilinogen and a dihydropyrazine rather than PBG. | ||
The lack of a simple route to PBG, while incongruous given the facile occurrence of the seemingly more complex steps 3 and 4 of the pathway, effectively crimps the entire process. The failure to find a viable structure-directed pathway to PBG has been referred to as the “Achilles’ heel” of the prebiogenesis of tetrapyrrole macrocycles.13,18 The resulting impasse leads to two opposing views concerning the origin of uroporphyrinogen in the prebiotic era: (i) uroporphyrinogen formed via structure-directed pathways through use of precursors other than PBG,13,18 or (ii) uroporphyrinogen did not form until a later era when appropriate catalysts (e.g., enzymes) were present to direct conversion of ALA to PBG. A parallel possibility is that diverse abiotic porphyrinogens – distinct from (and prebiotic predecessors to) uroporphyrinogen – formed in a structure-directed manner prior to the emergence of catalysts.
An investigation of the chemical foundation for the latter possibility revealed a new process that encompasses two of the distinct reaction steps (2 and 3) shown in Fig. 2: ALA and a 4-methoxy-substituted β-ketoester (2) react to yield a porphobilinogen analogue (3) that self-condenses to give the corresponding porphyrinogen (4, Fig. 6). The reaction proceeds under plausible prebiotic conditions [anaerobic reaction of ALA (1–5 mM) with 2 (2–40 mM) in water (pH 5–7) at 70–100 °C for >6 h]; upon oxidation the porphyrin is obtained in up to 10% yield.36 The 1,3-dicarbonyl motif directs enamine formation to the meso-carbon (C2), whereas the 4-methoxymethyl unit is untouched and becomes the α-methoxymethyl unit that equips the pyrrole for self-condensation. The resulting porphyrinogen and porphyrin are not found in contemporary living organisms; however, the success of this route provided a conceptual foundation for addressing the uroporphyrinogen problem. Here we describe a structure-directed route to uroporphyrinogen that entails reaction of acyclic species in anaerobic aqueous solution under mild conditions of temperature and pH. The same route with a different precursor affords coproporphyrinogen without the intermediacy of uroporphyrinogen, its contemporary biosynthetic precursor.
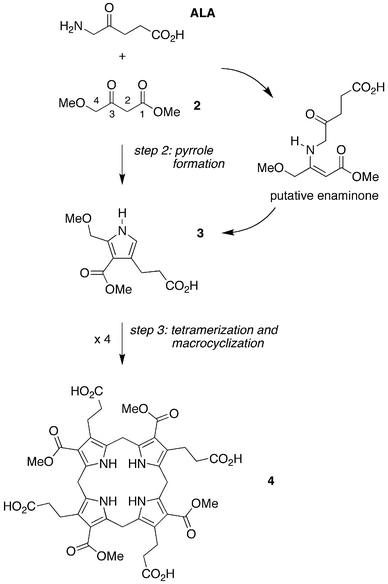 | ||
| Fig. 6 Integration of steps 2 and 3: hybrid condensation of ALA and a 4-methoxy-β-ketoester (2) affords pyrrole 3 equipped for self-condensation to give the non-biological porphyrinogen 4. | ||
Results and discussion
Uroporphyrinogen
The conceptual approach we investigated to form a PBG analogue from acyclic materials is shown in Fig. 7. A 2,4-diketone (1-AcOH) was chosen for reaction with ALA. Compound 1-AcOH is a hybrid of 5-methoxylevulinic acid and the simple 2,4-diketone acetylacetone. As a 5-methoxylevulinic acid species, 1-AcOH bears a methoxyacetyl unit at the 3-position. As an acetylacetone species, 1-AcOH bears methoxy groups at the two terminal methyl groups and an acetic acid substituent at the meso-carbon (C3). Thus, 1-AcOH is an ALA analogue yet differs in that 1-AcOH (i) does not self-condense; (ii) is expected to form the enamine wherein the carbon–carbon double bond encompasses the mesocarbon (3-position), and thereby direct carbon–carbon bond formation at the 3-γ positions; (iii) contains an α-methoxyacetyl unit that is lost upon pyrrole formation; and (iv) contains a methoxy substituent destined to be the leaving group at the α-methylpyrrole site upon reaction with ALA. The resulting pyrrole (MeOPBG-U) is an analogue of PBG that is equipped for self-condensation, bears the same β-substituents as PBG, and differs only in the presence of an α-methoxymethyl group rather than an α-aminomethyl unit.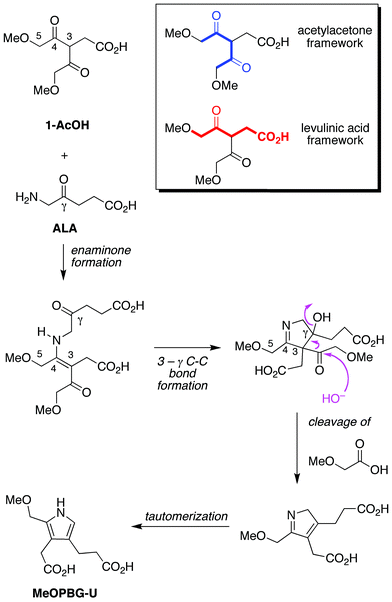 | ||
| Fig. 7 Compound 1-AcOH is a hybrid of the 2,4-diketone acetylacetone and the monoketone levulinic acid. Reaction with ALA is expected to preferentially yield the enaminone whereupon 3-γ bond formation closes the ring. Loss of the sacrificial methoxyacetyl unit (upon attack of HO−, H2O, or other nucleophile) followed by tautomerization affords the porphobilinogen analogue MeOPBG-U. Other pathways and products are possible (vide infra). | ||
The anaerobic reaction of ALA and 1-AcOH (120 mM each) at 60 °C and pH 5 for 24 h yielded the uroporphyrinogen [ESI-MS analysis gave m/z = 837.2873, (M + H)+, Fig. S1†] as shown in Fig. 8. The uroporphyrinogen was accompanied by a small amount of uroporphyrin (owing to background oxidation). Also present upon ESI-MS examination was a peak attributed to the formation of a pyrrolevia the Fischer-Fink pathway, termed MeO-FF-U [m/z = 314.1234, (M + H)+; Table S1†]; vide infra. Treatment of an aliquot of the reaction mixture with iodine in aqueous acid, a standard technique developed for conversion of uroporphyrinogen to uroporphyrin,31,33 enabled spectrophotometric quantitation on the basis of the characteristic Soret absorption band (Fig. 9A). Extraction of the acidic oxidized mixture with cyclohexanone gave uroporphyrin in crude form, which upon dissolution in neutral aqueous solution gave Soret and visible absorption bands (Fig. 9B) as well as fluorescence emission and excitation spectra (Fig. 9C), all of which were almost identical with that of an authentic sample of uroporphyrin (isomer III). ESI-MS analysis gave the uroporphyrin molecular ion [m/z = 831.2350, (M + H)+, Fig. S1†]. HPLC analysis with mass or absorption detection of the crude uroporphyrin gave three peaks, two of which exhibited retention times identical with those of commercially available samples of two uroporphyrin isomers (I and III) (Fig. 9D). Note that chromatographic conditions have only been developed for separation of three of the four uroporphyrin isomers.37 The ratio of uroporphyrin isomers observed from reaction of ALA and 1-AcOH resembled the equilibrium distribution obtained upon non-enzymic condensation of PBG (Fig. 9D).31
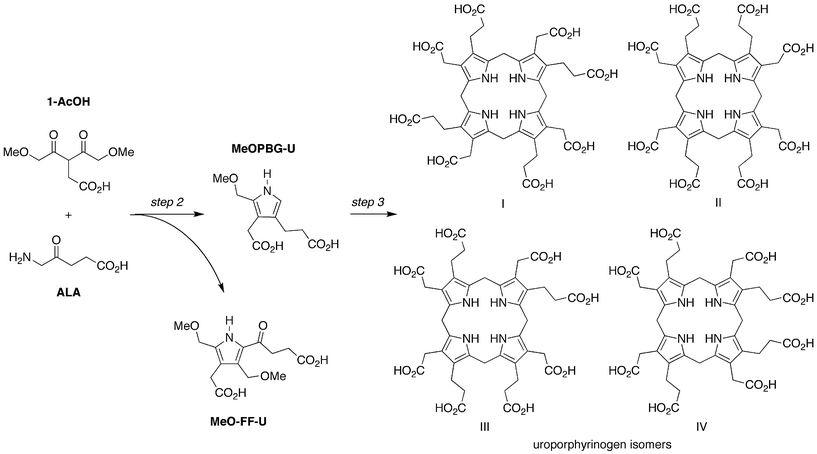 | ||
| Fig. 8 Condensation of ALA and 1-AcOH affords a porphobilinogen analogue (MeOPBG-U) that self-condenses to give uroporphyrinogen isomers (all four are shown). The competing Fischer-Fink pathway affords a fully substituted pyrrole (MeO-FF-U). | ||
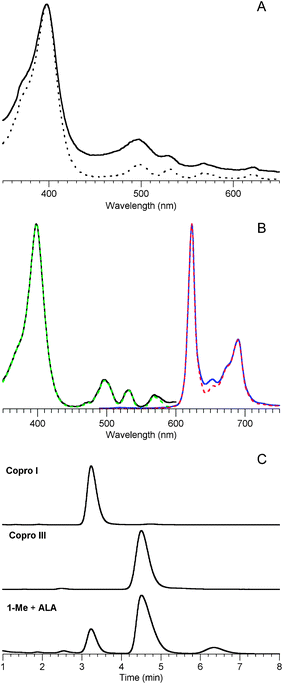
![Characterization data for the uroporphyrin product obtained from reaction of 1-AcOH and ALA (120 mM each, pH 5, 60 °C, 24 h). (A) Absorption spectra in 0.1 M HCl. An aliquot from the reaction mixture diluted 850-fold into a 2.6 mL cuvette (solid blue line) shows a trace of porphyrin (Soret band at 405 nm) and dipyrrin species (490 nm) owing to background oxidation. Upon addition of excess I2 to the cuvette (followed by discharging with Na2S2O3) the colorless porphyrinogen30 is converted to the porphyrin (solid black line). The spectrum of an authentic sample of uroporphyrin III is included for comparison (dotted red line, normalized at the Soret band of the crude sample). (B) Absorption spectra in 0.1 M sodium phosphate (pH 7). The crude uroporphyrin extracted from acidic brine (solid black line) is compared with an authentic sample of uroporphyrin III (dotted red line). (C) Fluorescence excitation (λem 678 nm) and emission spectra (λexc 398 nm) in 0.1 M sodium phosphate (pH 7) of the crude uroporphyrin (solid lines) are compared with those of an authentic sample of uroporphyrin III (dashed lines, normalized at the peak band of the crude sample). (D) HPLC with mass spectral detection [m/z = 831 ± 2, (M + H)+] of the crude uroporphyrin (showing three putative isomers) is compared with data from authentic samples of uroporphyrin I and uroporphyrin III. The chromatogram from the acidic condensation (90 °C, 1 h, air oxidation) of PBG is shown in overlay. Slight variation in retention times (e.g., 6 s at 4.4 min) occurred for a given sample on multiple runs. See Materials and methods for HPLC conditions.](/image/article/2011/NJ/c0nj00716a/c0nj00716a-f9.gif) | ||
| Fig. 9 Characterization data for the uroporphyrin product obtained from reaction of 1-AcOH and ALA (120 mM each, pH 5, 60 °C, 24 h). (A) Absorption spectra in 0.1 M HCl. An aliquot from the reaction mixture diluted 850-fold into a 2.6 mL cuvette (solid blue line) shows a trace of porphyrin (Soret band at 405 nm) and dipyrrin species (490 nm) owing to background oxidation. Upon addition of excess I2 to the cuvette (followed by discharging with Na2S2O3) the colorless porphyrinogen30 is converted to the porphyrin (solid black line). The spectrum of an authentic sample of uroporphyrin III is included for comparison (dotted red line, normalized at the Soret band of the crude sample). (B) Absorption spectra in 0.1 M sodium phosphate (pH 7). The crude uroporphyrin extracted from acidic brine (solid black line) is compared with an authentic sample of uroporphyrin III (dotted red line). (C) Fluorescence excitation (λem 678 nm) and emission spectra (λexc 398 nm) in 0.1 M sodium phosphate (pH 7) of the crude uroporphyrin (solid lines) are compared with those of an authentic sample of uroporphyrin III (dashed lines, normalized at the peak band of the crude sample). (D) HPLC with mass spectral detection [m/z = 831 ± 2, (M + H)+] of the crude uroporphyrin (showing three putative isomers) is compared with data from authentic samples of uroporphyrin I and uroporphyrin III. The chromatogram from the acidic condensation (90 °C, 1 h, air oxidation) of PBG is shown in overlay. Slight variation in retention times (e.g., 6 s at 4.4 min) occurred for a given sample on multiple runs. See Materials and methods for HPLC conditions. | ||
The reaction of ALA and 1-AcOH was examined under a range of conditions spanning concentration (5–240 mM), time (6–96 h), temperature (25–85 °C), and pH (5–7). Illustrative examples are as follows.
The reaction at 60 °C for 24 h afforded yields that increased from <0.5% to 7.9% (pH 5) or 10.7% (pH 7) upon going from dilute (5 mM) to concentrated (240 mM) reactions, with yields peaked in the 120–170 mM region (Fig. 10).
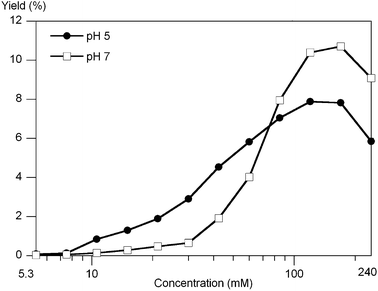 | ||
| Fig. 10 The yield of uroporphyrin as a function of the concentration of 1-AcOH and ALA (equimolar) upon reaction for 24 h at 60 °C at pH 5 or pH 7 determined upon oxidation of reaction aliquots. The data points are positioned at factors of 2−1/2 from the highest concentration (240 mM) examined. | ||
The reaction for 72 h at 60 °C gave uroporphyrin in 10.6% (120 mM, pH 7) or 4.3% yield (120 mM, pH 5).
The reaction at 25 °C (120 mM, pH 7) afforded uroporphyrin in 0.9% and 4.1% yield after 1 and 4 days, respectively, whereas at 85 °C for 6 h the yield was 2.2%.
In summary, while the interplay of concentration, pH, temperature and time are not yet fully delineated, a broad range of plausible early-Earth conditions38 was found to give successful condensation with yields of up to 10% of uroporphyrin. These results constitute the first laboratory demonstration of the structure-directed formation of uroporphyrinogen from acyclic reactants.
Coproporphyrinogen, a biosynthetically advanced tetrapyrrole macrocycle
The scope was examined by reaction of a substrate other than 1-AcOH under conditions identical to those to form uroporphyrinogen (Fig. 11). Thus, the reaction of the decarboxy analogue of 1-AcOH (1-Me) gave a molecular ion [m/z = 661.3236, (M + H)+] consistent with coproporphyrinogen (Fig. S2†). Use of LC-ESI-MS revealed the presence of four peaks as expected for the four possible coproporphyrinogen isomers (Fig. 12). Examination of the crude reaction mixture also revealed the presence of the Fischer-Fink pyrrole MeO-FF-C [m/z = 292.1161, (M + Na)+; Table S1†]; vide infra.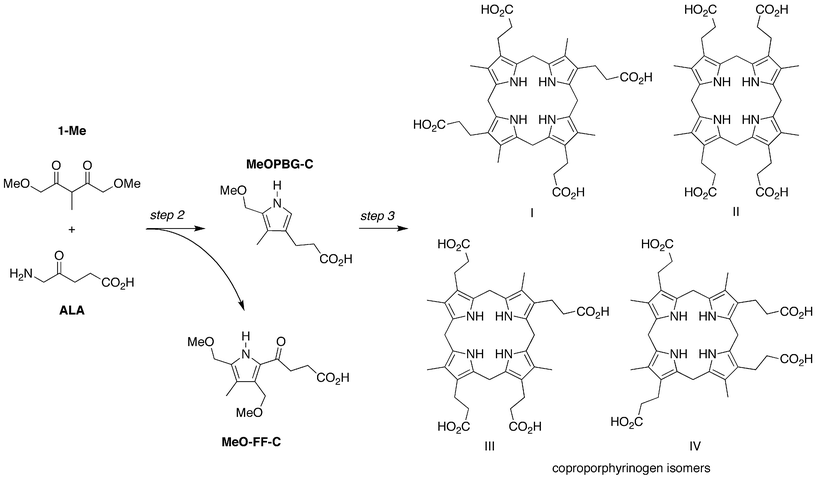 | ||
| Fig. 11 Condensation of 1-Me and ALA provides direct access to coproporphyrinogen isomers. The competing Fischer-Fink pathway affords a fully substituted pyrrole (MeO-FF-C). | ||
![LC-ESI-MS analysis of a sample from the reaction of 1-Me and ALA with detection at the mass channelm/z 659.4–663.1 shows four putative coproporphyrinogen isomers [(M + H)+]. See Materials and methods for HPLC conditions.](/image/article/2011/NJ/c0nj00716a/c0nj00716a-f12.gif) | ||
| Fig. 12 LC-ESI-MS analysis of a sample from the reaction of 1-Me and ALA with detection at the mass channelm/z 659.4–663.1 shows four putative coproporphyrinogen isomers [(M + H)+]. See Materials and methods for HPLC conditions. | ||
Oxidation of the reaction mixture obtained from 1-Me and ALA gave coproporphyrin in 10.6% overall yield (upon reaction at 120 mM, pH 7, 60 °C, 24 h). The absorption spectrum, fluorescence excitation spectrum, and fluorescence emission spectrum were nearly identical with those from an authentic sample of coproporphyrin III (Fig. 13A,B). Characterization of the coproporphyrin sample by mass spectrometry gave the expected molecular ion, and LC-ESI-MS showed three coproporphyrin isomers (Fig. S3†). HPLC with absorption detection also showed three coproporphyrin isomers (Fig. 13C). The same reaction at 25 °C for 4 days afforded coproporphyrin in 3.1% yield, whereas at 85 °C for 6 h the yield was 6.4%.
 | ||
| Fig. 13 Data for coproporphyrin obtained upon oxidation of a sample from the reaction of 1-Me and ALA (pH 7, 60 mM ALA and 120 mM 1-Me, 24 h, 60 °C). (A) Absorption spectra in n-propanol of the crude porphyrin extract (solid line) and comparison with the spectrum of an authentic sample of coproporphyrin III (dashed line); the spectra are normalized at the Soret band. (B) Fluorescence excitation (λem 678 nm) and emission spectra (λexc 398 nm) in n-propanol of the crude coproporphyrin (solid lines) obtained upon oxidation and extraction are compared with an authentic sample of coproporphyrin III (dashed lines, normalized at the peak bands). (C) HPLC with absorption spectral detection of the crude porphyrin extract compared with authentic samples of coproporphyrin I and coproporphyrin III. Three putative coproporphyrin isomers are observed in the crude sample. See Materials and methods for HPLC conditions. | ||
It is noteworthy that the pyrrole (MeOPBG-C) obtained in the reaction of 1-Me and ALA is not equipped to form a lactone with the β-alkylcarboxylic acid, as can occur with the pyrrole (MeOPBG-U) derived from 1-AcOH and ALA; with PBG;31 and perhaps with an α-(methoxymethyl)pyrrole used in a chemical synthesis of coproporphyrin39 (Fig. 14). Thus, the formation of such a lactone intermediate and/or intramolecular catalysis by the β-acetic acid unit is not essential to promote the condensation in step 3 (tetramerization and macrocyclization). Whether there are reaction conditions that proceed beneficially via a lactone intermediate remains to be determined.
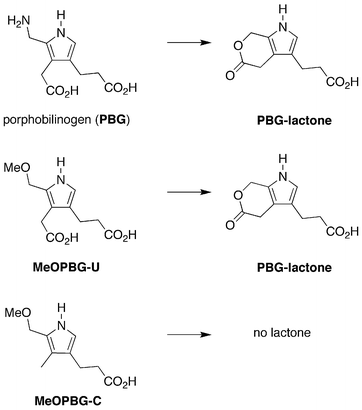 | ||
| Fig. 14 Possible lactone formation in step 3 (tetramerization and macrocyclization). | ||
Competing pathways to pyrroles
The reaction of an α-aminoketone and a β-diketone can proceed via multiple pathways, including here the desired Knorr and undesired Fischer-Fink routes (Fig. 15). Indeed, examination of a variety of reactions of ALA and 1-AcOH (or 1-Me) by ESI-MS revealed the presence of a peak due to the porphyrinogen (derived from the porphobilinogen-like Knorr pyrrole) as well as a peak due to the Fischer-Fink pyrrole (Table S1†). The Knorr and Fischer-Fink routes have their branch point at the step where the initial enaminone undergoes cyclization.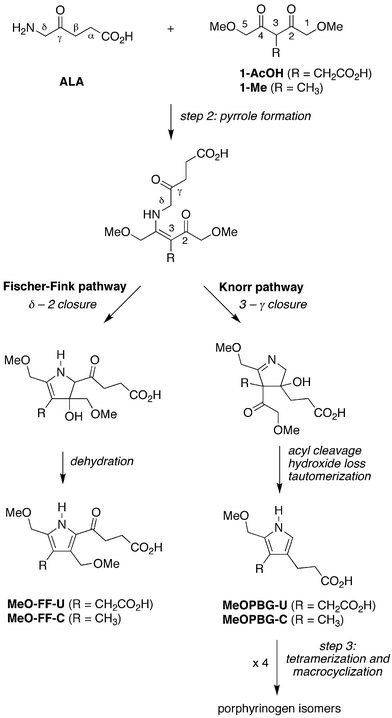 | ||
| Fig. 15 Competing Fischer-Fink and Knorr routes on the pathway to porphyrinogens. | ||
The Fischer-Fink route stems from condensation at the δ-methylene of ALA with the 2-carbonyl of 1-AcOH (or 1-Me) followed by dehydration. The Fischer-Fink pyrrole (MeO-FF-U or MeO-FF-C) is expected to be relatively unreactive owing to the deactivating effect of the α-acyl substituent. The Knorr pathway entails condensation of the enaminone C3-carbon with the ALA γ-ketone to yield the pyrroline intermediate (see Fig. 7). Because the enaminone nucleophile is a tertiary carbon, cleavage of the methoxyacetyl unit at the resulting quaternary carbon is required (followed by loss of hydroxide and tautomerization) to give the pyrrole equipped for self-condensation (MeOPBG-U or MeOPBG-C). Thus, one methoxyacetyl unit becomes an integral part of the structure (of MeOPBG-U or MeOPBG-C) while the second serves only as a temporary aide to direct enamine formation at the C3 site (Fig. 7). Other pyrroles are possible (e.g., by cleavage of the succinyl moiety along the Fischer-Fink route) but were not observed upon ESI-MS analysis of the reaction mixtures. Isomers of the Fischer-Fink pyrroles formed by 5 - γ closure (rather than δ - 2 closure as shown in Fig. 15) cannot be discriminated by nominal mass analysis, and while considered to be less likely, cannot at present be ruled out.
Butler and George carried out the reaction of ALA and 3-methylpentan-2,4-dione (5-Me), a substrate closely resembling the core structure of 1-AcOH or 1-Me.40 The Fischer-Fink pyrrole (6-Me) was the only solid product isolated, albeit in 16.7% yield.40 Were the Fischer-Fink pathway the only pathway available, of course, no porphyrinogen would result upon reaction of ALA and 1-AcOH or 1-Me. Because the result of Butler and George taken on face value indicated the reaction of ALA and 1-AcOH or 1-Me would not lead to porphyrinogen, we repeated the reaction of ALA and 3-substituted pentan-2,4-diones to assess the products under the exact conditions employed here that led to porphyrinogens. The 3-substituted pentan-2,4-diones lack the 1,5-dimethoxy substituents of 1-AcOH or 1-Me and hence are arrested with regards to subsequent condensations (Fig. 16). To assess the products unbiased by chemical separation methods, we employed ESI-MS analysis of the crude reaction mixtures (a measurement method not commonly available at the time when Butler and George performed their study).
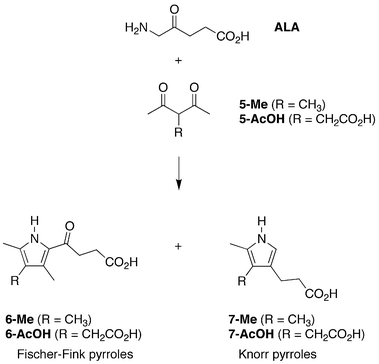 | ||
| Fig. 16 Competing Fischer-Fink and Knorr routes with 3-substituted pentan-2,4-diones. | ||
The reaction of ALA and 3-methylpentan-2,4-dione (5-Me, 60 mM) at 60 °C and pH 5 for 24 h upon ESI-MS analysis showed peaks for both the Fischer-Fink pyrrole 6-Me and the Knorr pyrrole 7-Me (Table S2†). Identical reaction of ALA and 3-acetyllevulinic acid (5-AcOH) showed peaks for both the Fischer-Fink pyrrole 6-AcOH and the Knorr pyrrole 7-AcOH. Whether the isolation of pyrrole 6-Me and not also 7-Me by Butler and George reflects relative yield or relative stability of the two pyrroles remains to be determined. In summary, the competing formation of the Fischer-Fink and Knorr pyrroles appears to be intrinsic to the reaction of a 3-substituted 2,4-diketone with ALA. Quantitation of the two pathways and examination of simple catalysts or conditions that might suppress the Fischer-Fink shunt require further study.
Outlook
The prior impasse concerning plausible prebiotic routes to tetrapyrrole macrocycles stemmed from inability to form the pyrrole PBG or equivalent thereof. Numerous routes are known for the chemical synthesis of pyrroles,41 including PBG;42–45 however, none employs relatively simple substances that spontaneously give rise to a pyrrole that contains all of the essential features of PBG. PBG (i) is water-soluble owing to the two carboxylic acid side chains and the aminomethyl unit, (ii) reacts as a 1° electrophile (or azafulvene equivalent) at the α-methyl site, and (iii) undergoes electrophilic substitution at the open α-site, a process potentiated by the two activating β-alkyl groups. Together, such features enable self-condensation of PBG in aqueous solution under mild conditions.The reaction pathway identified herein to form PBG analogues is fundamentally different from both the (efficient) enzymatic transformation and the (failed) solution dimerization of two molecules of ALA. The enzyme porphobilinogen synthase is believed to direct an aldol condensation (to set the critical β–γ bond, Fig. 5) between two molecules of ALA followed by imine formation to give the 5-membered ring.46 The dimerization of ALA in solution, which presumably entails initial formation of the imine, is thwarted by the greater reactivity of the δ versus β site for aldol condensation.34 The ALA-dimerization problem has been sidestepped here through a hybrid reaction of ALA with a 2,4-diketone bearing a 3-substituent and 1,5-dimethoxy units; one keto moiety functions sacrificially to direct reaction at the 3-position (corresponding to the β-site in ALA, which is comparatively unreactive in solution) and then is lost upon pyrrole formation. Other sacrificial directing groups (e.g., cyano) at the 3-position might serve equally well. The simplicity, structure-directed transformations, mild conditions, and non-negligible yields for the multistep process identified herein would appear to meet many criteria for a plausible prebiotic process.
On the other hand, one criterion that has not yet been met is a prebiotic route to 1-AcOH (or 1-Me), or for that matter to ALA. Ultimately, a claim to prebiotic relevance for the proposed route is contingent on substantiation of routes to such precursors from early Earth-available substances. With regards to the β-diketones (1-AcOH, 1-Me), β-dicarbonyl compounds are central to fatty acid biosynthesis/degradation and microbial polyketide synthesis.47 In addition, the α-methoxyacetyl unit is employed in polyketide synthesis.48 While it may never be possible to prove the occurrence of a prebiotic process, at a minimum, the study herein may reveal “indispensable chemical requirements that restrict the range of boundary conditions compatible with a chemistry of biogenesis.”49 In this context, the process highlights a number of issues central to studies of the origin of life.
A key issue concerns proto-metabolismversus extant metabolism, which de Duve has emphasized may be broadly parallel yet have distinct features.14 The following points deserve comment in this regard.
• The macrocycles uroporphyrinogen and coproporphyrinogen are ‘generationally simple yet architecturally complex’49 and form spontaneously from acyclic reactants; however, structure-directed reactions in general are far slower than enzymatically catalyzed processes.50 Here, the obvious limitations of the structure-directed processes include the competing formation of the undesired Fischer-Fink pyrrole, the formation of four porphyrinogen isomers, the requirement for somewhat high concentrations, and the comparatively low yields. It remains unknown how/when processes dictated solely by the intrinsic molecular structure of reactants gave way to more efficient and selective catalyzed reaction processes.
• Uroporphyrin, derived oxidatively from uroporphyrinogen, has been proposed as an ideal prebiotic photosensitizer,9–11,18,22,23 as has coproporphyrin.10 Both can now be readily formed from acyclic reactants yet neither is a constituent of extant biochemistry.4,19
• Coproporphyrinogen is a more advanced intermediate than uroporphyrinogen along the extant biosynthetic path to hemes and chlorophylls. The facile formation of coproporphyrinogen without uroporphyrinogen as a precursor raises questions concerning how/why the extant biosynthesis originated with reliance on ALA as the sole precursor followed by decarboxylation of the acetic acid side chains of uroporphyrinogen, versus employing two distinct substrates to directly form coproporphyrinogen.
• The yields to date are not high, but tetrapyrrole macrocycles persist for hundreds of millions of years in anaerobic environments,51 and hence would be expected to accumulate under prebiotic conditions.
The demonstration of a route from acyclic materials to porphyrinogens opens the door to the development of a chemical model for the prebiogenesis of tetrapyrrole macrocycles. Ultimately, such a model must begin with simple, early Earth-available precursors to ALA and 1-AcOH (or analogues) and integrate multiple successive transformations (steps 1-5, Fig. 2). Concerning step 1, we note that both ALA and 1-AcOH are of structural complexity intermediate between that of accepted prebiotic species such as amino acids and nucleosides. Steps 2 and 3 have been demonstrated herein, and precedent exists for step 4 (albeit as a stand-alone process).30 Toward greater molecular complexity, the reaction of diverse analogues of ALA and of 1-AcOH alone or in combinatorial processes, as well as in situ modification processes (step 5: side-chain alteration, reduction, metalation), are expected to yield diverse biotic and abiotic macrocycles encompassing a range of polarity and functionality.
The availability of tetrapyrrole macrocyclesvia model prebiotic processes enables examination of a variety of catalytic and energy-utilization processes central to origin-of-life problems. For example, the question of whether the earliest cells52 were chemotrophic or phototrophic has animated the life sciences since the time of Oparin and Haldane. The facile formation of diverse tetrapyrrole macrocycles from simple precursors may provide an opportunity to explore systematically the de novo emergence of proto-photosynthetic processes.
Materials and methods
Reactants
ALA is commercially available. The syntheses of compounds 1-AcOH and 1-Me (by alkylation of the known 1,5-dimethoxypentan-2,4-dione53) and 5-AcOH will be reported elsewhere.Reactions
All experiments were carried out at 0.5 mL scale under anaerobic conditions following techniques that have been described in detail for the reaction of ALA and 2.36 Reactions were performed in 0.5 M sodium acetate (pH 5) or 0.5 M 3-(N-morpholinyl)propylsulfonic acid (pH 7), respectively, with pH values determined at 25 °C.Porphyrin yields
The yield of porphyrin was determined by addition of a reaction aliquot (50 μL of a 30 mM reaction) to 0.1 M HCl (2.6 mL) in a cuvette under aerobic conditions, to which was then added 56 μL of I2 solution (50 mM in ethanol); subsequent treatment with 112 μL of Na2S2O3 (100 mM in water) discharged excess iodine. The absorption spectrum was quantitated using εSoret = 505![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.33 The oxidation process was scaled linearly for reactions at other concentrations, and for oxidations at larger scale. Treatment of the oxidized reaction mixture with acidic brine (3.6 M NaOAc, pH 3.2), extraction into cyclohexanone,54 and concentration of the organic extract afforded the crude porphyrin mixture.
000 M−1 cm−1.33 The oxidation process was scaled linearly for reactions at other concentrations, and for oxidations at larger scale. Treatment of the oxidized reaction mixture with acidic brine (3.6 M NaOAc, pH 3.2), extraction into cyclohexanone,54 and concentration of the organic extract afforded the crude porphyrin mixture.
HPLC conditions
HPLC of the uroporphyrin sample with mass spectral detection was carried out at 50 °C on a C18 column (50 × 2.1 mm, 2.7 μm particles) with the following solvents [A = aqueous NH4OAc (1.0 M, pH 5.15); B = 98% CH3CN, 2% water, and 0.3% formic acid; the CH3CN itself contained 2% water] and flow rate 0.35 mL min−1. The elution regimen consisted of 7% B for 4 min followed by transition to 10% B over 6 min. See Fig. 9D.HPLC of the coproporphyrinogen sample with mass spectral detection was carried out at 50 °C on a C18 column (50 × 2.1 mm, 2.7 μm particles) with the following solvents [A = 98% water and 2% CH3CN; B = 98% CH3CN, 2% water, and 0.3% formic acid; the CH3CN itself contained 2% water] and flow rate 0.4 mL min−1. The elution regimen consisted of 25% B for 2 min, transition to 75% B over 8 min, then transition to 25% B over 3 min. See Fig. 12.
HPLC of the coproporphyrin sample with absorption spectral detection (λdet = 405 nm) was carried out at 25 °C on a C18 column (100 × 2.1 mm, 5 μm particles) in isocratic mode (flow rate 0.8 mL min−1) using 26% of CH3CN and 74% of aqueous NH4OAc (1.0 M, pH 5.15). See Fig. 13C.
Fluorescence spectroscopy
The fluorescence spectra were obtained at room temperature with excitation and emission slit widths of 1.5 nm, and a sweep rate of 1 nm/2 s. The spectra were corrected for excitation intensity and instrument response on emission. The fluorescence emission spectra were obtained upon excitation at the peak of the Soret band with samples having absorbance values <0.06 in 1 cm pathlength cuvettes. Fluorescence excitation spectra were obtained upon emission detection at the Q(0,1) peak of the same samples.Mass spectrometry
High resolution exact mass measurements of the porphyrins and porphyrinogens were carried out using electrospray ionization (ESI) time-of-flight on an Agilent Technologies 6224 LC-TOF mass spectrometer operated in positive-ion mode (Duke University, with assistance of Dr George Dubay). High resolution exact mass measurements of the pyrroles were carried out using electrospray ionization (ESI) time-of-flight on an Agilent Technologies 6210 LC-TOF mass spectrometer operated in positive-ion mode (NC State University, with assistance of Ms. Danielle Lehman).Acknowledgements
This work was supported by a grant from the NSF Chemistry of Life Processes Program (NSF CHE-0953010). JSL wishes to thank Dr David C. Mauzerall for critical encouragement over many years concerning this “Achilles’ heel” of porphyrin evolution.13,18 The journal cover art accompanying this article was prepared by Ms. Margot Geist (www.geistlight.com) using a seascape image obtained at Cape Perpetua, Oregon.References
- S. L. Miller, in The Molecular Origins of Life, ed. A. Brack, Cambridge University Press, Cambridge, UK, 1998, pp. 59–85 Search PubMed.
- H. Gaffron, in Rhythmic and Synthetic Processes in Growth, ed. D. Rudnick, Princeton University Press, Princeton, NJ, USA, 1957, pp. 127–154 Search PubMed.
- S. Granick, Ann. N. Y. Acad. Sci., 1957, 69, 292–308 CrossRef CAS.
- S. Granick, in Evolving Genes and Proteins, ed. V. Bryson and H. J. Vogel, Academic Press, New York, USA, 1965, pp. 67–88 Search PubMed.
- J. D. Bernal, The Origin of Life, The World Publishing Company, Cleveland, OH, USA, 1967 Search PubMed.
- A. A. Krasnovsky, Origins Life, 1974, 5, 397–404 CrossRef CAS.
- A. A. Krasnovsky, Origins Life, 1976, 7, 133–143 CrossRef CAS.
- D. Mauzerall, Philos. Trans. R. Soc. London, Ser. B, 1976, 273, 287–294 CrossRef CAS.
- J. A. Mercer-Smith and D. Mauzerall, Photochem. Photobiol., 1981, 34, 407–410 CAS.
- J. A. Mercer-Smith and D. C. Mauzerall, Photochem. Photobiol., 1984, 39, 397–405 CrossRef CAS.
- J. A. Mercer-Smith, A. Raudino and D. C. Mauzerall, Photochem. Photobiol., 1985, 42, 239–244 CrossRef CAS.
- J. M. Olson and B. K. Pierson, Origins Life Evol. Biosphere, 1987, 17, 419–430 CrossRef CAS.
- D. C. Mauzerall, Origins Life Evol. Biosphere, 1990, 20, 293–302 CrossRef CAS.
- C. de Duve, in Blueprint for a Cell: The Nature and Origin of Life, Neil Patterson Publishers, Carolina Biological Supply Company, Burlington, NC, USA, 1991 Search PubMed.
- D. Mauzerall, Photosynth. Res., 1992, 33, 163–170 CrossRef CAS.
- M. T. Madigan, J. M. Martinko and J. Parker, in Brock Biology of Microorganisms, Prentice Hall, Upper Saddle River, NJ, USA, Eighth edn, 1997, p. 612 Search PubMed.
- D. W. Deamer, Microbiol. Mol. Biol. Rev., 1997, 61, 239–261 CAS.
- D. C. Mauzerall, Clin. Dermatol., 1998, 16, 195–201 CrossRef CAS.
- R. J. Porra, Photochem. Photobiol., 1997, 65, 492–516 CrossRef CAS.
- S. Granick, Harvey Lectures, 1950, 44, 220–245 Search PubMed.
- D. Mauzerall, in The Photosynthetic Bacteria, ed. R. K. Clayton and W. R. Sistrom, Plenum Press, New York, USA, 1978, pp. 223–231 Search PubMed.
- D. Mauzerall, in Bioorganic Chemistry, ed. E. E. van Tamelen, Academic Press, New York, USA, 1978, vol. IV, pp. 303–314 Search PubMed.
- S. Granick, in Biochemistry of Chloroplasts, ed. T.W. Goodwin, Academic Press, New York, USA, 1967, vol. 2, pp. 373–410 Search PubMed.
- P. A. Carapellucci and D. Mauzerall, Ann. N. Y. Acad. Sci., 1975, 244, 214–238 CrossRef CAS.
- J. Feitelson and D. Mauzerall, J. Phys. Chem., 1996, 100, 7698–7703 CrossRef CAS.
- D. Mauzerall, J. Phys. Chem., 1962, 66, 2531–2533 CrossRef CAS.
- D. Mauzerall, J. Am. Chem. Soc., 1962, 84, 2437–2445 CrossRef CAS.
- T. K. With, Biochem. J., 1975, 147, 249–251 CAS.
- J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300–311 CrossRef CAS.
- D. Mauzerall and S. Granick, J. Biol. Chem., 1958, 232, 1141–1162 CAS.
- D. Mauzerall, J. Am. Chem. Soc., 1960, 82, 2605–2609 CAS.
- R. B. Frydman, S. Reil and B. Frydman, Biochemistry, 1971, 10, 1154–1160 CrossRef CAS.
- D. Mauzerall, J. Am. Chem. Soc., 1960, 82, 2601–2605 CAS.
- A. R. Butler and S. George, Tetrahedron, 1992, 48, 7879–7886 CrossRef CAS.
- A. R. Chaperon, H. Bertschy, A.-L. Franz-Schrumpf, B. Hugelet, A. Neels, H. Stoeckli-Evans and R. Neier, Chimia, 2003, 57, 601–606 CrossRef CAS.
- J. S. Lindsey, M. Ptaszek and M. Taniguchi, Origins Life Evol. Biosphere, 2009, 39, 495–515 CrossRef CAS.
- J. M. Rideout, D. J. Wright and C. K. Lim, J. Liq. Chromatogr. Relat. Technol., 1983, 6, 383–394 CrossRef CAS.
- J. F. Kasting and M. T. Howard, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 1733–1742 CrossRef CAS.
- I. T. Kay, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 901–905 CrossRef CAS.
- A. R. Butler and S. D. George, Tetrahedron, 1993, 49, 7017–7026 CrossRef CAS.
- G. P. Bean, in Pyrroles, Part 1, ed. R. A. Jones, John Wiley & Sons, Inc., New York, USA, 1990, pp. 105–294 Search PubMed.
- P. Bobal and R. Neier, Trends Org. Chem., 1997, 6, 125–144 Search PubMed.
- A. R. Chaperon, T. M. Engeloch and R. Neier, Angew. Chem., Int. Ed., 1998, 37, 358–360 CrossRef CAS.
- R. Neier, J. Heterocycl. Chem., 2000, 37, 487–508 CrossRef CAS.
- C. P. Soldermann, R. Vallinayagam, M. Tzouros and R. Neier, J. Org. Chem., 2008, 73, 764–767 CrossRef CAS.
- E. K. Jaffe, Bioorg. Chem., 2004, 32, 316–325 CrossRef CAS.
- S. Smith and S.-C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC.
- Y. A. Chan, A. M. Podevels, B. M. Kevany and M. G. Thomas, Nat. Prod. Rep., 2009, 26, 90–114 RSC.
- A. Eschenmoser, Tetrahedron, 2007, 63, 12821–12844 CrossRef CAS.
- R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS.
- H. J. Callot and R. Ocampo, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, USA, 2000, vol. 1, pp. 349–398 Search PubMed.
- T. Cavalier-Smith, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 969–1006 CrossRef CAS.
- A. H. Alberts and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 3545–3553 CrossRef CAS.
- C. Rimington and A. Benson, Biochem. J., 1967, 105, 1085–1090 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectral data for the porphyrinogens, porphyrins, and various pyrroles. See DOI: 10.1039/c0nj00716a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |