Structural characteristics that make chlorophylls green: interplay of hydrocarbon skeleton and substituents
Olga
Mass
a,
Masahiko
Taniguchi
a,
Marcin
Ptaszek
a,
Joseph W.
Springer
b,
Kaitlyn M.
Faries
b,
James R.
Diers
c,
David F.
Bocian
*c,
Dewey
Holten
*b and
Jonathan S.
Lindsey
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina, 27695-8204, USA. E-mail: jlindsey@ncsu.edu
bDepartment of Chemistry, Washington University, St. Louis, Missouri, 63130-4889, USA. E-mail: holten@wustl.edu
cDepartment of Chemistry, University of California, Riverside, California, 92521-0403, USA. E-mail: David.Bocian@ucr.edu
First published on 10th November 2010
Abstract
Understanding the effects of substituents on natural photosynthetic pigments is essential for gaining a deep understanding of why such pigments were selected over the course of evolution for use in photosynthetic systems. This knowledge should provide for a more thoughtful design of artificial light-harvesting systems. The hydrocarbon skeleton of all chlorophylls is phorbine, which contains an annulated five-membered (isocyclic) ring in addition to the reduced pyrrole ring characteristic of chlorins. A phorbine and a 131-oxophorbine (which bears an oxo group in the isocyclic ring) were synthesized as benchmark molecules for fundamental spectral and photophysical studies. The phorbine and 131-oxophorbine macrocycles lack peripheral substituents other than a geminal dimethyl group in the reduced ring to stabilize the chlorin chromophore. The spectral properties and electronic structure of the zinc or free base 131-oxophorbine closely resemble those of the corresponding analogues of chlorophyll a. Accordingly, the fundamental electronic properties of chlorophylls are primarily a consequence of the 131-oxophorbine base macrocycle.
Introduction
Chlorophylls are the most famous members of the chlorin family.1 The chlorin designation stems from the presence of one reduced pyrrole ring in the tetrapyrrole macrocycle. However, the carbon skeleton of chlorophylls is not simply a cyclic tetrapyrrole, but also contains an annulated five-membered ring. This hydrocarbon framework is known as phorbine.2 A phorbine that carries an oxo group at the 131-position, as is the case with all chlorophylls, is known as a 131-oxophorbine. The structures of chlorin, phorbine, 131-oxophorbine, chlorophyll a and chlorophyllb are shown in Chart 1. Inspection of the chlorophyll structure leads naturally to questions concerning the requirements for the fifth ring, the given set of peripheral substituents, and the extent to which such features contribute to the distinct spectral and electronic properties of the chlorophylls.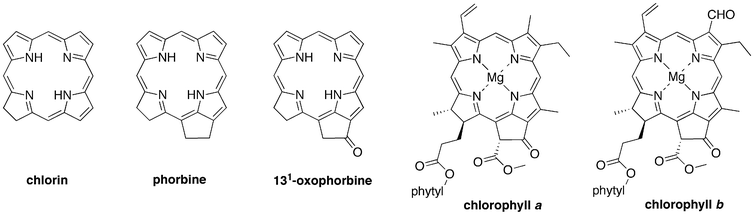 | ||
| Chart 1 Building the skeleton of chlorophyll a. | ||
The question of ‘why chlorophyll?’ has been asked a number of times from different perspectives. Mauzerall considered the features of chlorophyll with regards to the biosynthetic pathway, the electronic properties that give rise to spectral and photochemical properties, and how such properties are well matched for photosynthetic function.3 A generation later the same question was addressed from the vantage point of a deeper molecular understanding of the diverse roles of chlorophylls in photosynthetic energy- and electron-transfer processes, including the apparently unique role of chlorophyll a in oxygenic photosynthesis.4 Björn considered the spectral matching of the chlorophyll absorption spectrum with solar radiation5 and also the prospects for finding habitable exoplanets on the basis of chlorophyll spectral properties.6 The spectroscopic properties of chlorophylls have been studied in detail.7 Still, the role of the specific substituents about the perimeter of the tetrapyrrole macrocycle in engendering the characteristic features of chlorophylls has largely been unaddressed, at least in a systematic manner.
The presence of specific substituents at sites around the perimeter of the chlorophyll macrocycle can have a significant effect on the spectral, electronic, and photochemical properties. For example, the sole distinction between chlorophyll a and chlorophyllb is the presence of a 7-methylversus 7-formyl group, yet this structural change results in a significant difference in spectral properties. This difference is exploited in nature, where chlorophyllsa and b serve together to broaden spectral coverage in plant photosynthesis.8
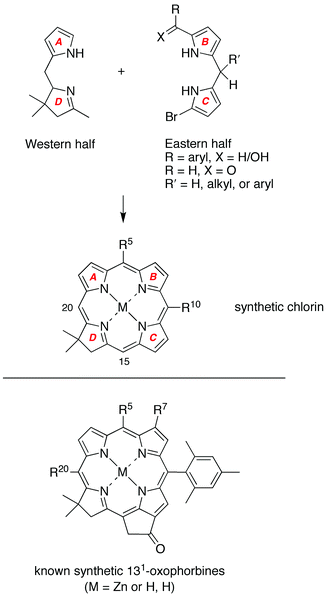
The traditional route to probe the effects of substituents in chlorophylls has been to modify intact chlorophylls.9–22 Such semisynthesis methods have resulted in structural changes such as hydrogenation of the 3-vinyl group, reduction of the 13-keto group, and insertion of other metals. However, achieving more global changes such as removing all alkyl groups (2, 7, 8, 12 positions) has not been feasible via semisynthesis. An alternative entails total synthesis, which has been used to prepare analogues of the naturally occurring chlorophylls. Our own work has been focused on the development of de novo synthetic methods for the preparation of stable synthetic analogues of the naturally occurring chlorophylls.23–43
Herein we report the use of recently developed synthetic methods to gain access to a series of benchmark analogues of chlorophylls that lack any substituents other than a stabilizing geminal dimethyl group in the reduced ring. The compounds include phorbine FbP, chlorin FbC–A13, and 131-oxophorbine FbOP (Chart 2), as well as their zinc chelates (ZnP, ZnC–A13, and ZnOP). The analogous chlorins FbC and ZnC have been synthesized previously,31 as has the 17-oxochlorin FbOC.32 The spectral and photophysical properties of each compound are also presented and are accompanied by density functional theory (DFT) calculations aimed at characterizing the frontier molecular orbitals. Collectively, these studies systematically address the issue of how structural modifications to the base chlorin macrocycle contribute to the electronic and spectral characteristics of chlorophylls.
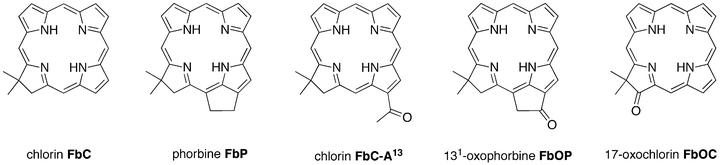 | ||
| Chart 2 Benchmark molecules for chlorophylls. | ||
Results and discussion
I Synthesis
 | ||
| Chart 3 Historical precedents in 131-oxophorbine and phorbine chemistry. | ||
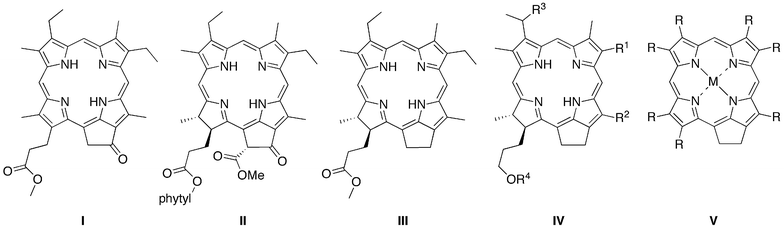
Each of the aforementioned syntheses of phorbines, dehydrophorbines, and 131-oxophorbines affords a macrocycle that contains a full complement of substituents at the β-pyrrole positions. The de novo synthesis of chlorins that we developed joins an Eastern half (rings B and C) and a Western half (rings A and D), where the latter contains a geminal methyl group in the pyrroline ring to stabilize the resulting chlorin macrocycle toward adventitious dehydrogenation (Scheme 1).23–43 The de novo route requires far more synthetic effort than semisynthetic methods that begin with chlorophylls,9–22 yet affords greater versatility in the scope and range of substituents that can be introduced. The available substituent pattern includes a benchmark chlorin (FbC) that differs from chlorin itself only in the presence of the geminal dimethyl group in the reduced ring.31 Methodology also has been developed to convert a synthetic 13-acetylchlorin to the corresponding 131-oxophorbine (via 15-bromination and intramolecular α-arylation).30 In this manner, seven free base 131-oxophorbines have been prepared, each of which bears a mesityl group at the 10-position and one or two other substituents. Four such 131-oxophorbines incorporate various substituents at the 7-position.30,41 Nevertheless, a synthesis of the phorbine and 131-oxophorbine lacking any meso or β-pyrrole substituents has heretofore not been accomplished. Access to such macrocycles is fundamentally important because these molecules serve as benchmarks against which to compare the spectral and photophysical properties of the full library of synthetic chlorins, as well as the naturally occurring chlorophylls.
 | ||
| Scheme 1 De novo synthesis of chlorins (top). All prior known synthetic 131-oxophorbines incorporate a 10-mesityl group and one or two other substituents (bottom). | ||
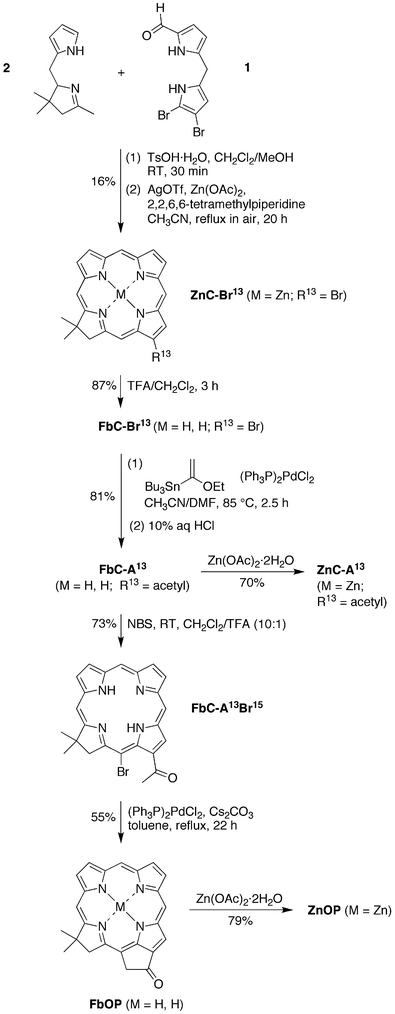 | ||
| Scheme 2 Synthesis of the 131-oxophorbines FbOP and ZnOP. | ||
Stille coupling
54 of FbC–Br13 with tributyl(1-ethoxyvinyl)tin and [(PPh3)2Pd]Cl2 in CH3CN/DMF (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶2)42 for 2.5 h at 85 °C followed by acidic workup afforded 13-acetylchlorin FbC–A13 in 81% yield. Regioselective 15-bromination of FbC–A13 was achieved with 1 molar equivalent of NBS under acidic conditions41 for 1 h at room temperature to give FbC–A13Br15 in 73% yield. Acidic conditions cause deactivation of ring B of the macrocycle and thus prevent bromination at the 7-position. Intramolecular α-arylation30,55,56 of the 13-acetyl group in the presence of (PPh3)2PdCl2 and Cs2CO3 in toluene installed the isocyclic ring (spanning positions 13 and 15) in 55% yield. Metalation of FbC–A13 and FbOP with Zn(OAc)2·2H2O gave ZnC–A13 and ZnOP in 70% and 79% yield, respectively. The zinc oxophorbine ZnOP was successfully purified by precipitation; however, other attempts to purify by column chromatography using 1% TEA resulted in complete decomposition.
∶2)42 for 2.5 h at 85 °C followed by acidic workup afforded 13-acetylchlorin FbC–A13 in 81% yield. Regioselective 15-bromination of FbC–A13 was achieved with 1 molar equivalent of NBS under acidic conditions41 for 1 h at room temperature to give FbC–A13Br15 in 73% yield. Acidic conditions cause deactivation of ring B of the macrocycle and thus prevent bromination at the 7-position. Intramolecular α-arylation30,55,56 of the 13-acetyl group in the presence of (PPh3)2PdCl2 and Cs2CO3 in toluene installed the isocyclic ring (spanning positions 13 and 15) in 55% yield. Metalation of FbC–A13 and FbOP with Zn(OAc)2·2H2O gave ZnC–A13 and ZnOP in 70% and 79% yield, respectively. The zinc oxophorbine ZnOP was successfully purified by precipitation; however, other attempts to purify by column chromatography using 1% TEA resulted in complete decomposition.
∶1) at 65 °C afforded crude tosylhydrazone–FbP, which upon reduction with sodium cyanoborohydride in the presence of TsOH·H2O in MeOH/THF gave tosylhydrazide–FbP. The latter was converted to the target phorbine FbP in 42% overall yield in EtOH/DMF for 5 h at 80 °C in the presence of sodium acetate trihydrate. Metalation of phorbine FbP with Zn(OAc)2·2H2O gave the zinc chelate phorbine ZnP in 90% yield (Scheme 3).
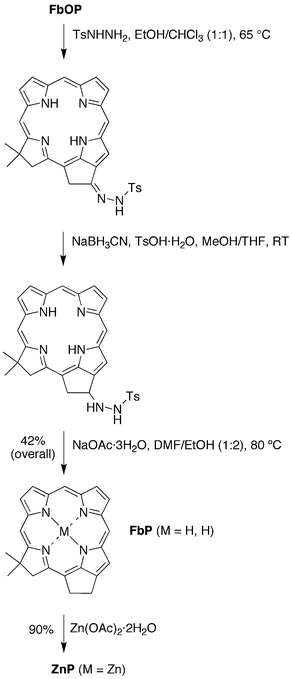 | ||
| Scheme 3 Synthesis of the free base phorbine FbP and the zinc phorbine ZnP. | ||
II Characterization
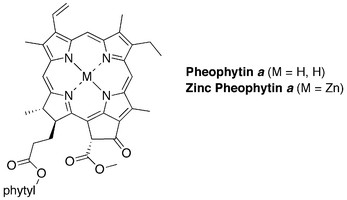 | ||
| Chart 4 Analogues of chlorophyll a. | ||
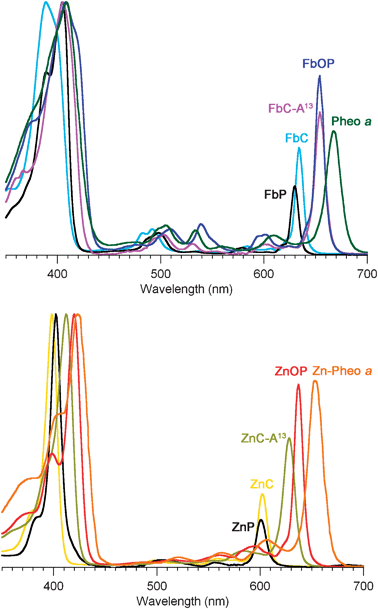 | ||
| Fig. 1 Absorption spectra of synthetic chlorins and phorbinesversuspheophytins75,76 (normalized at the B-band maxima). Top panel: free base macrocycles. Bottom panel: zinc chelates. The solvent is diethyl ether (Pheo aa and Zn–Pheo a) or toluene (all other compounds). | ||
The absorption spectrum of each compound is comprised of three main spectral regions. The strong red-region feature (600–670 nm) is the Qy(0,0) band; on average, this band lies ∼30 nm to longer wavelength for the free base compound versus the corresponding zinc chelate. The weaker features in the blue-green to yellow spectral region (470–580 nm) are the Qx bands; these features are somewhat stronger for the free base compounds than the zinc chelates. The intense bands in the near-ultraviolet region are the Bx and By features, also known as the Soret bands; these bands are generally superimposed for the zinc chelates but split for the free base compounds. The positions and relative intensities of the B and Qy bands for the various macrocycles are listed in Table 1.
| Compound | λ B [fwhm]/nm | Δ B b/cm−1 | λ Qy [fwhm]/nm | Δ Qy b/cm−1 | I B/IQyc | Σ Qy /ΣBd | HOMO–LUMOe/eV | Φ f f | τ s g/ns |
|---|---|---|---|---|---|---|---|---|---|
| a In toluene at room temperature unless noted otherwise. b The redshift of the band relative to the corresponding band of the parent chlorin (FbC or ZnC). c Ratio of the peak intensities of the B and Qy bands. d Ratio of the integrated intensities of the Qy manifold [Qy(0,0) and Qy(1,0) bands] and B manifold [Bx and By origins and first vibronic overtones]. e Energy gap between the HOMO and LUMO orbitals. f Fluorescence quantum yield (error ± 7%). g Lifetime of the singlet excited state measured using fluorescence techniques (error ± 8%). h Absorption data from ref. 75 (in diethyl ether). i Absorption data from ref. 76 (in diethyl ether). j Essentially the same value (2.65 eV) is obtained with an H2O axial ligand. k From ref. 34. | |||||||||
| FbC | 389 [33] | — | 633 [10] | — | 2.4 | 0.19 | 2.69 | 0.20 | 8.8 |
| FbP | 404 [30] | −950 | 629 [9] | 100 | 3.6 | 0.15 | 2.72 | 0.28 | 10.6 |
| FbOP | 408 [55] | −1170 | 654 [11] | −490 | 1.5 | 0.24 | 2.58 | 0.30 | 11.5 |
| FbC –A 13 | 405 [34] | −970 | 654 [12] | −500 | 1.8 | 0.24 | 2.59 | 0.24 | 7.8 |
| FbOC | 400 [27] | −710 | 634 [9] | −10 | 4.6 | 0.09 | 2.76 | 0.12 | 8.4 |
| Pheo aa h | 409 [53] | −1200 | 668 [17] | −810 | 2.0 | 0.25 | 2.47 | ||
| ZnC | 398 [13] | — | 602 [11] | — | 3.6 | 0.24 | 2.67 | 0.062 | 1.7 |
| ZnP | 402 [2] | −240 | 601 [12] | 11 | 4.6 | 0.27 | 2.69 | 0.064 | 1.7 |
| ZnOP | 419 [19] | −1230 | 637 [17] | −910 | 1.6 | 0.42 | 2.55 | 0.23 | 5.1 |
| ZnC – A 13 | 411 [21] | −760 | 627 [14] | −680 | 1.9 | 0.40 | 2.56 | 0.23 | 4.3 |
| ZnOC | 412 [14] | −810 | 602 [9] | −10 | 3.4 | 0.23 | 2.68 | 0.030 | 0.82 |
| Zn –Pheo a i | 423 [38] | −1480 | 653 [17] | −1300 | 1.4 | 0.40 | 2.43 | ||
| MgC | 402 [10] | — | 607 [12] | — | 4.3 | 0.28 | 2.63j | 0.26 | 6.9 |
| Chl aa | 432 [40] | −1730 | 665 [18] | −1440 | 1.3 | 0.63 | 2.40 | 0.33 | 6.3k |
The characteristics of the main absorption features (Bx, By, Qx, Qy) depend on the nature of the macrocycle and peripheral substituents. Pheo aa bears two major auxochromes, the 13-keto group (embedded in the isocyclic ring) and the 3-vinyl group. The 131-oxophorbines FbOP and ZnOP lack the 3-vinyl group; the phorbines FbP and ZnP additionally lack the 13-keto group; the chlorin FbC–A1313 further lacks the five-membered ring; the base chlorins FbC and ZnC lack any substituents (except for the geminal dimethyl group in the reduced pyrrole ring common to all of the synthetic compounds examined here). Of major interest is the manner in which the nature of the macrocycle influences the position and intensity of the Qy(0,0) band. This is so because this feature corresponds to excitation to the lowest singlet excited state, from which key energy- and electron-transfer events of photosynthesis are initiated. The salient points concerning the Qy(0,0) band are as follows:
(1) In the free base series, FbOP has the most intense Qy(0,0) band (relative to the Soret maximum). The Qy(0,0) band of FbC–A13 exhibits the same (largest) bathochromic shift as FbOP, but is about 80% as intense. Both compounds nearly mimic the spectrum of Pheo aa. In the zinc-chelate series, ZnOP has the relatively most intense Qy(0,0) band and the largest bathochromic shift, with ZnC–A13 second in the series in both categories.
(2) The spectral properties of phorbines (FbP and ZnP) closely resemble those of chlorins (FbC or ZnC). Both sets of compounds exhibit a Qy(0,0) band that lacks the intensity and bathochromic shift of the keto-substituted analogues.
(3) The main absorption characteristics of unsubstituted chlorin MgC33 closely resemble those of ZnC (Table 1). This similarity parallels that for the native magnesium-containing chlorophyll a (Chl aa) and the zinc-bearing analogue Zn–Pheo a (Table 1).
In summary, the chlorin chromophore alone affords a poor mimic of the spectral properties of chlorophylls. However, the addition of a 13-keto group to the chlorin is essential (and suffices) to closely mimic the absorption spectrum of the naturally occurring chlorin pigment; it is anticipated that the added effect of a 3-vinyl group would give a near complete match to the spectrum of chlorophyll a.
The fluorescence yield of the magnesium chlorin MgC is larger than those for the free base form FbC and zinc chelate ZnC (0.26, 0.20, 0.062) while the singlet excited-state lifetime is intermediate (6.9 ns, 8.8 ns, 1.7 ns). This trend follows those for the fluorescence yields (0.14, 0.09, 0.030) and excited-state lifetimes (13 ns, 8.9 ns, 2.1 ns) of magnesium, free base, and zinc tetraphenylporphyrins.77–79
The collective photophysical data show that the addition of a 13-keto group to the chlorin results in a more intense and bathochromically shifted Qy(0,0) absorption feature, accompanied by fluorescence yields and singlet excited-state lifetimes for the zinc chelates that are not nearly as diminished compared to the free base analogues as for the other macrocycles. The decreased excited-state energy and increased optical-transition strength affords better utilization of the red region of the solar spectrum. The 13-keto group also increases the excited-state lifetime, which is desirable for allowing a higher yield of the light-driven energy- or charge-transfer reactions of photosynthesis. The lengthening of the excited-state lifetime is unexpected in that the lower excited-state energy would normally (based on the energy-gap law80) lead to a shortening of the lifetime due to enhancement of the nonradiative (internal conversion) decay pathway. Thus, the incorporation of a 13-keto group in a chlorin or phorbine macrocycle results in a very favorable motif from a photophysical point of view.
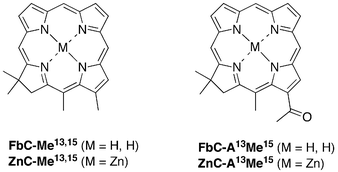 | ||
| Chart 5 Fictive chlorins for which DFT calculations were performed. | ||
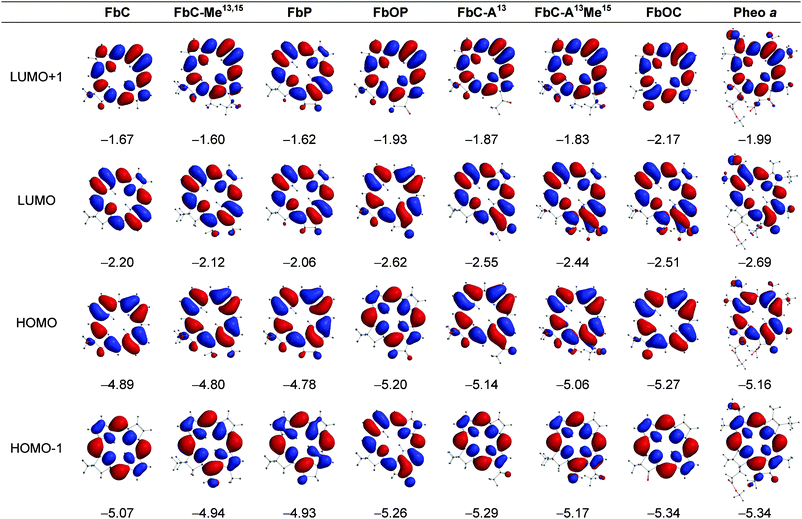 | ||
| Fig. 2 Electron-density distributions and energies (eV) of the frontier MOs of the free base compounds. | ||
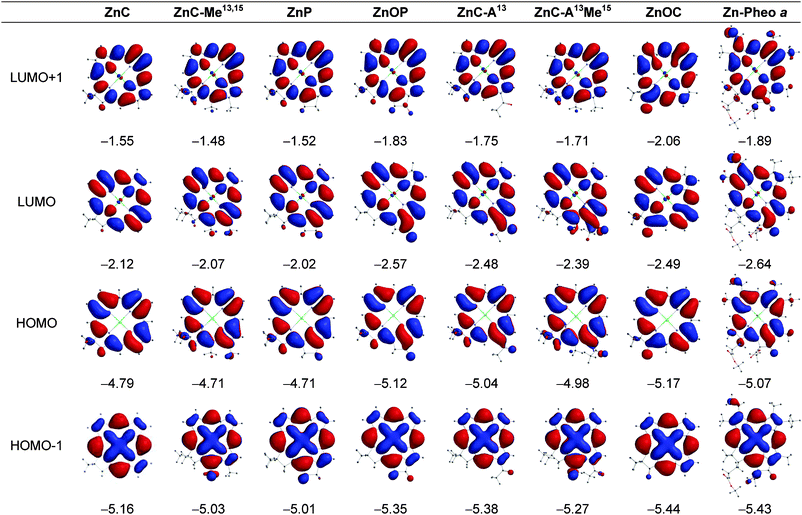 | ||
| Fig. 3 Electron-density distributions and energies (eV) of the frontier MOs of the zinc chelates. | ||
Macrocycle structure and MO energies. The key findings from the MO calculations are as follows:
(1) The conversion of the chlorin to a phorbine results in a small (0.08–0.14 eV) shift in the HOMO and LUMO energies to less negative values. Such shifts correspond to the phorbine being slightly easier to oxidize and slightly harder to reduce than the chlorin. This connection follows from our prior studies on about two dozen synthetic zinc chlorins that showed linear correlations between the calculated MO energies and measured redox potentials.34,35 Moreover, the calculations reported herein on the fictive chlorins FbC–Me13,1513,15 and ZnC–Me13,15 suggest that about half (free base) or almost the entire (zinc chelate) MO-energy shifts can be accounted for simply by the presence of the 13- and 15-substituents and that closure to the five-membered ring (and associated ring strain) has only a small effect.
(2) Incorporation of a 13-keto group in a chlorin to produce FbC–A13 or ZnC–A13 gives a 0.25 eV shift in the HOMO energy and a 0.35–0.36 eV shift in the LUMO energy to more negative values. The analogous incorporation of an oxo group in a phorbine to produce the 131-oxophorbine (e.g., FbP to FbOP) gives an even larger 0.41–0.42 eV shift in the HOMO energy and a larger still 0.55–0.56 eV shift in the LUMO energy to more negative values. The direction of the shifts is such that the presence of the 13-keto group will make the molecule harder to oxidize and easier to reduce. The calculations on the fictive chlorin ZnC–A13M15, together with those described above for ZnC–M13,15, suggest that closure to the five-membered ring (and associated ring strain) has a somewhat greater effect on the frontier-MO energies when the 13-keto group is present (e.g., FbP to FbOP) than in its absence (e.g., FbC to FbC–A13).
(3) The effect of the 13-keto group on the LUMO energy is larger than the effect on the HOMO energy by an average of 0.11 eV for the four cases (13-keto addition to chlorin or 131-oxo addition to phorbine and for free base and zinc forms). This effect can be understood by the generally larger electron density on the 13-keto group for the LUMO compared to the HOMO (Fig. 2 and 3). The unequal effect of the 13-keto group on the LUMO and HOMO alters the HOMO − LUMO energy gap and thus affects the energy/wavelength of the Qy(0,0) absorption band, as described further below.
(4) The 13-keto substituted chlorins FbC–A1313 and ZnC–A13 have HOMO energies that are only slightly (0.02–0.03 eV) less negative and LUMO energies that are modestly (0.14–0.16 eV) less negative than those of the corresponding natural pigments Pheo aa and Zn–Pheo a. Thus, the simple presence of a 13-keto group, without closure to the five-membered ring to give the oxophorbine, is sufficient to give a macrocycle which is slightly easier to oxidize and only modestly harder to reduce than the related photosynthetic pigment (i.e., Pheo aa). Carrying the comparison one step further, the unsubstituted oxophorbines FbOP and ZnOP have HOMO energies that are actually slightly (0.04–0.05 eV) more negative and LUMO energies that are slightly (0.07 eV) less negative than those of Pheo aa and Zn–Pheo a. Thus, the oxophorbine macrocycle encodes the primary electronic properties that dictate the redox potentials of the native photosynthetic pigments, with only small net adjustments due to the substituents at the six β-pyrrole positions.
Absorption spectra and MO energies. Fig. 4 (top) plots the energies of the HOMO, LUMO, HOMO-1, and LUMO+1 versusQy(0,0) absorption energy/wavelength (Table 1) for the various macrocycles, along with linear fits to the data and values indicating the slopes of the trend lines. For both the free base macrocycles (open symbols and dashed lines) and zinc chelates (closed symbols and solid lines), the LUMO exhibits a much greater dependence on macrocycle characteristics than the HOMO or other orbitals (HOMO-1 and LUMO+1). In particular, the slope of the trend lines encompassing the addition of a 13-keto group to a chlorin (e.g.FbC to FbC–A13) or a 131-oxo group to a phorbine (e.g.FbP to FbOP) is twice as large for the LUMO than for the other three frontier MOs.
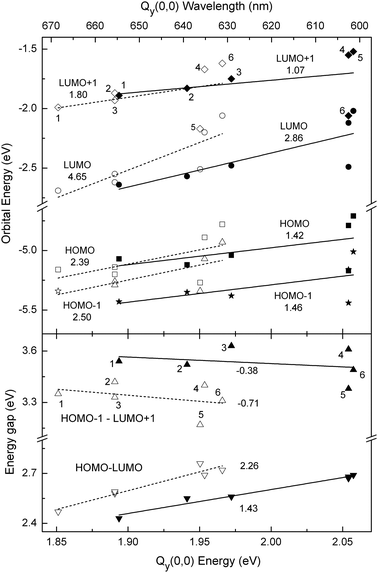 | ||
| Fig. 4 Calculated frontier MO energies (top panel) and energy gaps (bottom panel) versus the measured Qy(0,0) absorption-band energy (bottom axis) and wavelength (top axis) for free base macrocycles (open symbols) and zinc chelates (closed symbols). Top: energies LUMO+1 (diamonds), LUMO (circles), HOMO (squares), HOMO-1 (stars). Bottom: energy gaps HOMO-1 − LUMO+1 (up triangles), and HOMO − LUMO (down triangles). The indicated values are the slopes of the trend lines for free base macrocycles (dashed lines) and zinc chelates (solid lines). The points for the different macrocycles are labeled as follows: Pheo aa (1), FbC–A13 (2), FbOP (3), FbC (4), FbOC (5), and FbP (6), and similarly for the zinc chelates. | ||
Close inspection of the orbital energies listed in Fig. 2 and 3 shows that the trends depicted in Fig. 4 (top) reflect a correlation between a decrease in the Qy(0,0) absorption-band energy (an increase in wavelength) and a decrease in the HOMO − LUMO energy gap upon the presence of a 13-keto group (in either a chlorin or phorbine). This tracking of the Qy(0,0) energy/wavelength with the HOMO − LUMO energy gap is shown explicitly in Fig. 4 (bottom) for the free base macrocycles (open downward triangles and dashed line) and zinc chelates (closed downward triangles and solid line). The bottom panel of Fig. 4 also shows that, in contrast, the energy gap between the LUMO+1 and HOMO-1 increases slightly as the Qy(0,0) energy decreases (wavelength increases), and with a trend-line slope that is about one-third that of the HOMO −LUMO energy gap.
The collective results depicted in Fig. 4 show that (1) the bathochromic shift in the position of the Qy(0,0) absorption band is driven by a reduction in the HOMO − LUMO energy gap. (2) The reduction in the HOMO − LUMO energy gap is in turn driven by a stronger effect of the 13-keto substituent on the LUMO energy than the HOMO energy. (3) These effects can be understood by the greater electron density on the 13-keto group for the LUMO compared to the other frontier MOs.
The results above can be taken one step further by application of Gouterman's four-orbital model.82–84 Within this model, the energy/wavelength of the Qy(0,0) absorption band depends on the quantity ΔEavg defined in eqn (1), namely the average of the HOMO − LUMO and LUMO+1 − HOMO-1 energy gaps, whereas the intensity of the band depends on the quantity ΔEdif defined in eqn (2), namely the difference in these two orbital energy gaps:
| ΔEavg = [(HOMO-1 − LUMO+1) + (HOMO − LUMO)]/2 | (1) |
| ΔEdif = [(HOMO-1 − LUMO+1) − (HOMO − LUMO)] | (2) |
Fig. 5 (top) shows a good overall correlation between ΔEavg and the Qy(0,0) absorption energy/wavelength for both the free base macrocycles and zinc chelates. Fig. 5 (bottom) shows a good correlation between ΔEdif and the integrated intensity of the Qy(0,0) absorption band (Table 1), especially for the free base compounds (open symbols and dashed line). We have previously utilized the four-orbital model to understand how the macrocycle-substituent pattern controls the MO energies and the spectral characteristics of a large set of synthetic free base and zinc chlorins.34,35
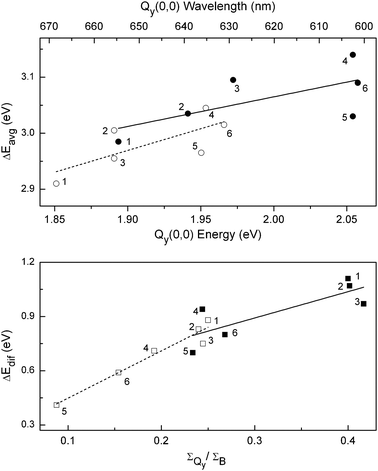 | ||
| Fig. 5 (top) Calculated average of the HOMO-1 − LUMO+1 and HOMO − LUMO energy gaps versus the measured Qy(0,0) absorption-band energy (bottom axis) and wavelength (top axis) for free base macrocycles (open symbols and dashed trend line) and zinc chelates (closed symbols and solid trend line). (bottom) Calculated difference in the HOMO-1 − LUMO+1 and HOMO − LUMO energy gaps versus the ratio of the integrated intensities of the Qy and B absorption manifolds for the free base macrocycles (open symbols and dashed trend line) and zinc chelates (closed symbols and solid trend line). The points for the different macrocycles are labeled as follows: Pheo aa (1), FbC–A13 (2), FbOP (3), FbC (4), FbOC (5), and FbP (6), and similarly for the zinc chelates. | ||
In the present study of unsubstituted parent macrocycles (chlorin, phorbine, oxophorbine), the use of the four-orbital framework (involving simple sums and differences of MO energy gaps) provides additional fundamental insights into the intimate relationship between the hydrocarbon skeleton, electronic structure, and characteristics of the long-wavelength absorption band of the natural photosynthetic pigments. Moreover, this framework and understanding has predictive value in aiding the design of synthetic analogues of the parent macrocycles and of the fully substituted photosynthetic pigments to allow better coverage of the solar spectrum for use in artificial systems for solar-energy conversion. The ability to mimic the spectral properties of chlorophylls with far simpler and synthetically accessible macrocycles augurs well for the use of such hydroporphyrins in a variety of artificial photosynthetic architectures.
Finally, we note that the studies here have employed zinc chelates rather than magnesium as is present in the chlorophylls. Compared with zinc, magnesium affords a bathochromic shift of the Qy band of about 10 nm, a 3–4-fold longer singlet excited-state lifetime, more facile ground-state oxidation, a more potent excited-state reductant, and tighter binding of oxygenic apical ligands. On the other hand, magnesium is more susceptible to demetalation. Any application would need to balance the advantageous photophysical properties of magnesium chlorinsversus the greater chemical stability of the zinc chelates.
Experimental section
I General synthetic methods
All 1H NMR (300 MHz and 400 MHz) and 13C NMR (100 MHz) spectra were obtained in CDCl3 unless noted otherwise. Mass spectra of chlorins were obtained vialaser desorption ionization mass spectrometry without a matrix or using a matrix of 1,4-bis(5-phenyloxazol-2-yl)benzene. Electrospray ionization mass spectrometry (ESI-MS) data are reported for the molecule ion or protonated molecule ion. Column chromatography was performed with flash silica. NBS was recrystallized (water). All other commercially available materials were used as received. Absorption and fluorescence spectra were obtained in toluene at room temperature unless noted otherwise. All of the Pd-mediated coupling reactions were carried out under argon using standard Schlenk-line procedures (e.g., three freeze–pump–thaw cycles were performed prior to and after the addition of the palladium reagent, for a total of six such cycles).∶1)] to afford a green solid (35 mg, 16%): 1H NMR (400 MHz, THF-d8) δ 2.06 (s, 6H), 4.63 (s, 2H), 8.72 (s, 1H), 8.80 (d, J = 4.1 Hz, 1H), 8.89 (s, 1H), 8.94–8.96 (m, 2H), 9.09 (d, J = 4.1 Hz, 1H), 9.15 (s, 1H), 9.58 (s, 1H), 9.64 (s, 1H); 13C NMR (THF-d8) δ 31.5, 46.4, 51.7, 94.0, 95.1, 109.08, 109.17, 115.8, 128.6, 129.4, 129.8, 133.0, 133.7, 144.6, 147.6, 147.8, 148.3, 149.3, 155.1, 159.8, 171.6; MALDI-MS obsd 479.9; ESI-MS obsd 481.00126 (M + H)+ corresponds to 479.99398 (M), calcd 479.99281 (M = C22H17BrN4Zn); λabs 401, 610 nm; λem 614 nm (λex 401 nm).
∶1)] afforded a purple solid (25 mg, 87%): 1H NMR (400 MHz, THF-d8) δ −2.45 (br, 2H), 2.06 (s, 6H), 4.70 (s, 2H), 9.015–9.017 (m, 2H), 9.06 (d, J = 4.4 Hz, 1H), 9.11 (s, 1H), 9.19 (s, 1H), 9.31 (d, J = 4.4 Hz, 1H), 9.39 (s, 1H), 9.82 (s, 1H), 9.89 (s, 1H); 13C NMR (THF-d8) δ 31.3, 47.4, 52.7, 95.0, 95.9, 106.9, 107.5, 112.4, 125.0, 128.8, 129.1, 133.5, 133.7, 133.9, 136.4, 136.9, 141.8, 153.1, 153.5, 164.1, 176.1; LD-MS obsd 417.9; ESI-MS obsd 419.08744 (M + H)+ corresponds to 418.08017 (M), calcd 418.07931 (M = C22H19BrN4); λabs 391, 641 nm.
∶1)] to afford a purple solid (37 mg, 81%): 1H NMR (300 MHz) δ −1.80 (br, 2H), 2.03 (s, 6H), 3.24 (s, 3H), 4.61 (s, 2H), 8.82 (s, 1H), 8.91 (d, J = 4.4 Hz, 1H), 8.92 (d, J = 4.2 Hz, 1H), 9.00 (d, J = 4.2 Hz, 1H), 9.14 (d, J = 4.4 Hz, 1H), 9.53 (s, 1H), 9.64 (s, 1H), 9.76 (s, 1H), 10.1 (s, 1H); 13C NMRδ 29.9, 31.2, 47.0, 51.9, 95.0, 97.7, 106.1, 109.5, 125.7, 128.9, 129.4, 130.4, 132.6, 134.0, 136.9, 137.3, 142.3, 151.6, 154.3, 165.0, 177.6, 197.0, 203.0; MALDI-MS obsd 381.9; ESI-MS obsd 383.18648 (M + H)+ corresponds to 382.1792 (M), calcd 382.1794 (M = C24H22N4O); λabs 405 (log ε = 4.91), 654 (log ε = 4.64) nm.
∶1)] to give a purple solid (30 mg, 73%): 1H NMR (300 MHz) δ −2.14 (br, 1H), −1.90 (br, 1H), 2.04 (s, 6H), 3.19 (s, 3H), 4.59 (s, 2H), 8.82 (s, 1H), 8.88 (d, J = 4.3 Hz, 1H), 8.91 (d, J = 5.0 Hz, 1H), 8.97 (d, J = 4.3 Hz, 1H), 9.13 (d, J = 5.0 Hz, 1H), 9.13 (s, 1H), 9.61 (s, 1H), 9.73 (s, 1H); 13C NMRδ 31.6, 34.8, 46.8, 55.2, 95.0, 95.7, 105.9, 109.7, 126.37, 126.46, 129.2, 130.5, 132.7, 133.8, 134.6, 136.9, 137.6, 141.5, 151.3, 155.0, 163.1, 177.4, 202.3; LD-MS obsd 460.0; ESI-MS obsd 461.09695 (M + H)+ corresponds to 460.08968 (M); calcd 460.08987 (M = C24H21BrN4O); λabs 399, 664 nm.
∶1)] to afford a green solid (5 mg, 70%): 1H NMR (400 MHz, THF-d8) δ 2.02 (s, 6H), 3.08 (s, 3H), 4.55 (s, 2H), 8.58 (s, 1H), 8.73 (d, J = 4.4 Hz, 1H), 8.85 (d, J = 4.4 Hz, 1H), 8.93 (d, J = 4.4 Hz, 1H), 9.01 (d, J = 4.4 Hz, 1H), 9.48 (s, 1H), 9.60 (s, 1H), 9.63 (s, 1H), 9.87 (s, 1H); MALDI-MS 444.2; ESI-MS obsd 445.09909 (M + H)+ corresponds to 444.09181 (M), calcd 444.09286 (M = C24H20N4OZn); λabs 412, 628 nm; λem 632 nm (λex 412 nm).
∶1)] to afford a purple solid (12 mg, 55%): 1H NMR (300 MHz) δ −2.23 (br, 1H), −0.17 (br, 1H), 1.98 (s, 6H), 4.18 (s, 2H), 5.03 (s, 2H), 8.64 (s, 1H), 8.73 (d, J = 4.3 Hz, 1H), 8.81 (d, J = 4.3 Hz, 1H), 8.85–8.87 (m, 1H), 9.91 (s, 1H), 9.03–9.06 (m, 1H), 9.37 (s, 1H), 9.50 (s, 1H); 13C NMRδ 31.1, 47.8, 48.57, 48.74, 95.1, 103.8, 106.1, 111.1, 115.6, 126.8, 130.3, 132.5, 132.8, 134.3, 138.1, 138.9, 143.1, 149.2, 152.6, 154.9, 157.3, 177.1, 195.8; MALDI-MS obsd 380.0; ESI-MS obsd 381.17088 (M + H)+ corresponds to 380.16360 (M); calcd 380.16371 (M = C24H20N4O); λabs 408 (log ε = 4.74), 654 (log ε = 4.53) nm; λem 657 nm (λex 408 nm).
∶1, 6 mL) was treated with Zn(OAc)2·2H2O (95 mg, 0.43 mmol). The reaction mixture was stirred for 20 h at room temperature. CH2Cl2 was added, and the mixture was concentrated. The resulting solid was dissolved in CH2Cl2, and the resulting organic solution was washed (saturated aqueous NaHCO3, water), dried (Na2SO4), and concentrated to afford a green solid. The solid was washed with hexanes three times and then was dissolved in the minimal amount of CH2Cl2. A double volume of hexanes subsequently was added, which afforded a precipitate that was isolated by centrifugation (10 mg, 78%): 1H NMR (400 MHz, THF-d8) δ 2.05 (s, 6H), 4.34 (s, 2H), 5.04 (s, 2H), 8.59 (s, 1H), 8.78–8.80 (m, 2H), 8.95 (d, J = 4.4 Hz, 1H), 9.01, (d, J = 4.2 Hz, 1H), 9.05 (s, 1H), 9.39 (s, 1H), 9.74 (s, 1H); LD-MS obsd 442.1; ESI-MS obsd 443.08407 (M + H)+ corresponds to 442.07679; calcd 442.07587 (C24H18N4OZn); λabs 420, 637 nm; λem 640 (λex 420 nm).
∶1) was treated with tosylhydrazide (44 mg, 0.24 mmol) at 65 °C. The reaction mixture was stirred for 2 h at 65 °C and then 2 h at room temperature. The reaction mixture was concentrated, and the solid was washed three times [H2O/methanol (1∶4)] and dried under high vacuum. The crude solid was dissolved in THF (4 mL), and the resulting solution was treated with sodium cyanoborohydride (22 mg, 0.32 mmol) followed by a solution of TsOH·H2O (48 mg, 0.25 mmol) in MeOH (1 mL). After 2 h, the reaction mixture was diluted with CH2Cl2 and quenched by the addition of saturated aqueous NaHCO3. The organic phase was washed with water, dried over Na2SO4, and filtered. The filtrate was concentrated to afford a green solid. The solid was dissolved in DMF/EtOH (5 mL, 1∶2), and the solution was treated with sodium acetate trihydrate (54 mg, 0.40 mmol). After heating at 80 °C for 5 h, the mixture was allowed to cool to room temperature, whereupon CH2Cl2 and water were added. The organic phase was washed (water and brine), dried (Na2SO4) and concentrated. Column chromatography (silica, CH2Cl2/hexanes [1∶1]) afforded a green solid (12 mg, 42%): 1H NMR (400 MHz) δ −3.42 (br, 1H), −1.67 (br, 1H), 2.10 (s, 6H), 4.17–4.19 (m, 2H), 4.46 (s, 2H), 4.78–4.80 (m, 2H), 8.79 (s, 1H), 9.04–9.06 (m, 2H), 9.08 (d, J = 4.1 Hz, 1H), 9.16 (d, J = 4.1 Hz, 1H), 9.31–9.33 (m, 1H), 9.73 (s, 1H), 9.98 (s, 1H); 13C NMRδ 25.9, 31.9, 37.6, 47.4, 49.9, 95.1, 103.4, 105.8, 112.3, 116.2, 122.6, 127.4, 131.1, 132.5, 133.5, 139.2, 143.0, 145.2, 147.9, 149.2, 152.7, 160.1, 171.4; LD-MS obsd 366.1; ESI-MS obsd 367.19219 (M + H)+ corresponds to 366.18492 (M), calcd 366.18445 (M = C24H22N4); λabs 405 (log ε = 5.16), 630 (log ε = 4.59) nm.
II Photophysical measurements
Static absorption (Varian Cary 100) and fluorescence (Spex Fluorolog Tau 2 or PTI Quantamaster 40) measurements were performed as described previously.73,74Argon-purged solutions of the samples in toluene with an absorbance of ≤0.10 at the excitation wavelength were used for the fluorescence spectral, quantum yield, and lifetime measurements. Fluorescence lifetimes were obtained using a phase modulation technique and Soret-band excitation74 (Spex Fluorolog Tau 2) or via decay measurements using excitation pulses at 337 nm from a nitrogen laser and time-correlated-single-photon-counting detection (Photon Technology International LaserStrobe TM-3). Emission measurements employed 2–4 nm excitation- and detection-monochromator bandwidths and 0.2 nm data intervals. Emission spectra were corrected for detection-system spectral response. Fluorescence quantum yields were determined relative to chlorophyll a in benzene (Φf = 0.325)85 or chlorophyll a in toluene, which was found here to have the same value as in benzene.III Density functional theory calculations
DFT calculations were performed with Spartan '08 for Windows (Wavefunction, Inc.) on a PC (Dell Optiplex GX270) equipped with a 3.2 GHz CPU and 3 GB of RAM.86 The hybrid B3LYP functional and the 6-31G* basis set were employed. The equilibrium geometries were fully optimized using the default parameters of the Spartan '08 program. A methyl group was substituted for the phytyl chain in the calculations on Pheo aa and Zn–Pheo a.Acknowledgements
This work was supported by grants from the Division of Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences of the U.S. Department of Energy to D.F.B. (DE-FG02-05ER15660), D.H. (DE-FG02-05ER15661) and J.S.L. (DE-FG02-96ER14632). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation.References
- H. Scheer, in Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, 2006, vol. 25, pp. 1–26 Search PubMed.
- G. P. Moss, Pure Appl. Chem., 1987, 59, 779–832.
- D. Mauzerall, Ann. N. Y. Acad. Sci., 1973, 206, 483–494 CrossRef CAS.
- L. O. Björn, G. C. Papageorgiou, R. E. Blankenship and Govindjee, Photosynth. Res., 2009, 99, 85–98 CrossRef.
- L. O. Björn, Photosynthetica, 1976, 10, 121–129.
- L. O. Björn, G. C. Papageorgiou, D. Dravins and Govindjee, Curr. Sci., 2009, 96, 1171–1175 CAS.
- M. Kobayashi, M. Akiyama, H. Kano and H. Kise, in Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, 2006, vol. 25, pp. 79–94 Search PubMed.
- W. L. Butler, in The Chlorophylls, ed. L. P. Vernon and G. R. Seely, Academic Press, New York, 1966, pp. 343–379 Search PubMed.
- W. Flitsch, Adv. Heterocycl. Chem., 1988, 43, 73–126 CAS.
- P. H. Hynninen, in Chlorophylls, ed. H. Scheer, CRC Press, Boca Raton, FL, 1991, pp. 145–209 Search PubMed.
- K. M. Smith, in Chlorophylls, ed. H. Scheer, CRC Press, Boca Raton, FL, 1991, pp. 115–143 Search PubMed.
- F.-P. Montforts, B. Gerlach and F. Höper, Chem. Rev., 1994, 94, 327–347 CrossRef CAS.
- F.-P. Montforts and M. Glasenapp-Breiling, Prog. Heterocycl. Chem., 1998, 10, 1–24 Search PubMed.
- L. Jaquinod, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 1, pp. 201–237 Search PubMed.
- M. G. H. Vicente, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 1, pp. 149–199 Search PubMed.
- R. K. Pandey and G. Zheng, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 6, pp. 157–230 Search PubMed.
- F.-P. Montforts and M. Glasenapp-Breiling, Fortschr. Chem. Org. Naturst., 2002, 84, 1–51 CAS.
- V. Y. Pavlov and G. V. Ponomarev, Chem. Heterocycl. Compd. (N. Y.), 2004, 40, 393–425 CrossRef CAS.
- M. O. Senge, A. Wiehe and C. Ryppa, in Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Advances in Photosynthesis and Respiration, Springer, Dordrecht, The Netherlands, 2006, vol. 25, pp. 27–37 Search PubMed.
- S. Fox and R. W. Boyle, Tetrahedron, 2006, 62, 10039–10054 CrossRef CAS.
- M. Galezowski and D. T. Gryko, Curr. Org. Chem., 2007, 11, 1310–1338 CrossRef CAS.
- A. M. G. Silva and J. A. S. Cavaleiro, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, Amsterdam, 2008, vol. 19, pp. 44–69 Search PubMed.
- J.-P. Strachan, D. F. O'Shea, T. Balasubramanian and J. S. Lindsey, J. Org. Chem., 2000, 65, 3160–3172 CrossRef CAS.
- T. Balasubramanian, J.-P. Strachan, P. D. Boyle and J. S. Lindsey, J. Org. Chem., 2000, 65, 7919–7929 CrossRef CAS.
- M. Taniguchi, D. Ra, G. Mo, T. Balasubramanian and J. S. Lindsey, J. Org. Chem., 2001, 66, 7342–7354 CrossRef CAS.
- M. Taniguchi, H.-J. Kim, D. Ra, J. K. Schwartz, C. Kirmaier, E. Hindin, J. R. Diers, S. Prathapan, D. F. Bocian, D. Holten and J. S. Lindsey, J. Org. Chem., 2002, 67, 7329–7342 CrossRef CAS.
- M. Taniguchi, M. N. Kim, D. Ra and J. S. Lindsey, J. Org. Chem., 2005, 70, 275–285 CrossRef CAS.
- M. Ptaszek, J. Bhaumik, H.-J. Kim, M. Taniguchi and J. S. Lindsey, Org. Process Res. Dev., 2005, 9, 651–659 Search PubMed.
- (a) J. K. Laha, C. Muthiah, M. Taniguchi, B. E. McDowell, M. Ptaszek and J. S. Lindsey, J. Org. Chem., 2006, 71, 4092–4102 CrossRef CAS; (b) J. K. Laha, C. Muthiah, M. Taniguchi, B. E. McDowell, M. Ptaszek and J. S. Lindsey, J. Org. Chem., 2009, 74, 5122 CrossRef CAS.
- J. K. Laha, C. Muthiah, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2006, 71, 7049–7052 CrossRef CAS.
- M. Ptaszek, B. E. McDowell, M. Taniguchi, H.-J. Kim and J. S. Lindsey, Tetrahedron, 2007, 63, 3826–3839 CrossRef CAS.
- M. Taniguchi, M. Ptaszek, B. E. McDowell and J. S. Lindsey, Tetrahedron, 2007, 63, 3840–3849 CrossRef CAS.
- M. Taniguchi, M. Ptaszek, B. E. McDowell, P. D. Boyle and J. S. Lindsey, Tetrahedron, 2007, 63, 3850–3863 CrossRef CAS.
- H. L. Kee, C. Kirmaier, Q. Tang, J. R. Diers, C. Muthiah, M. Taniguchi, J. K. Laha, M. Ptaszek, J. S. Lindsey, D. F. Bocian and D. Holten, Photochem. Photobiol., 2007, 83, 1110–1124 CrossRef CAS.
- H. L. Kee, C. Kirmaier, Q. Tang, J. R. Diers, C. Muthiah, M. Taniguchi, J. K. Laha, M. Ptaszek, J. S. Lindsey, D. F. Bocian and D. Holten, Photochem. Photobiol., 2007, 83, 1125–1143 CrossRef CAS.
- C. Muthiah, J. Bhaumik and J. S. Lindsey, J. Org. Chem., 2007, 72, 5839–5842 CrossRef CAS.
- K. E. Borbas, V. Chandrashaker, C. Muthiah, H. L. Kee, D. Holten and J. S. Lindsey, J. Org. Chem., 2008, 73, 3145–3158 CrossRef CAS.
- C. Muthiah, M. Ptaszek, T. M. Nguyen, K. M. Flack and J. S. Lindsey, J. Org. Chem., 2007, 72, 7736–7749 CrossRef CAS.
- C. Muthiah, H. L. Kee, J. R. Diers, D. Fan, M. Ptaszek, D. F. Bocian, D. Holten and J. S. Lindsey, Photochem. Photobiol., 2008, 84, 786–801 CrossRef CAS.
- C. Ruzié, M. Krayer and J. S. Lindsey, Org. Lett., 2009, 11, 1761–1764 CrossRef CAS.
- C. Muthiah, D. Lahaye, M. Taniguchi, M. Ptaszek and J. S. Lindsey, J. Org. Chem., 2009, 74, 3237–3247 CrossRef CAS.
- O. Mass, M. Ptaszek, M. Taniguchi, J. R. Diers, H. L. Kee, D. F. Bocian, D. Holten and J. S. Lindsey, J. Org. Chem., 2009, 74, 5276–5289 CrossRef CAS.
- M. Ptaszek, D. Lahaye, M. Krayer, C. Muthiah and J. S. Lindsey, J. Org. Chem., 2010, 75, 1659–1673 CrossRef CAS.
- H. Fischer and O. Laubereau, Justus Liebigs Ann. Chem., 1938, 535, 17–37 CrossRef CAS.
- H. Fischer and A. Oestreicher, Justus Liebigs Ann. Chem., 1940, 546, 49–57 CAS.
- H. Fischer and H. Siebel, Justus Liebigs Ann. Chem., 1932, 499, 84–108 CrossRef CAS.
- H. Wolf, Justus Liebigs Ann. Chem., 1966, 695, 98–111 CrossRef CAS.
- H. Brockmann Jr. and R. Tacke-Karimdadian, Liebigs Ann. Chem., 1979, 419–430 CrossRef.
- T. D. Lash, R. P. Balasubramaniam, J. J. Catarello, M. C. Johnson, D. A. May Jr., K. A. Bladel, J. M. Feeley, M. C. Hoehner, T. G. Marron, T. H. Nguyen, T. J. Perun Jr., D. M. Quizon, C. M. Shiner and A. Watson, Energy Fuels, 1990, 4, 668–674 CrossRef CAS.
- B. Zhang and T. D. Lash, Tetrahedron Lett., 2003, 44, 7253–7256 CrossRef CAS.
- T. D. Lash, W. Li and D. M. Quizon-Colquitt, Tetrahedron, 2007, 63, 12324–12342 CrossRef CAS.
- G. W. Kenner, J. Rimmer, K. M. Smith and J. F. Unsworth, J. Chem. Soc., Perkin Trans. 1, 1978, 845–852 RSC.
- C. Bauder, R. Ocampo and H. J. Callot, Tetrahedron, 1992, 48, 5135–5150 CrossRef CAS.
- M. Kosugi, T. Sumiya, Y. Obara, M. Suzuki, H. Sano and T. Migita, Bull. Chem. Soc. Jpn., 1987, 60, 767–768 CAS.
- H. Muratake and M. Natsume, Tetrahedron Lett., 1997, 38, 7581–7582 CrossRef CAS.
- H. Muratake, M. Natsume and H. Nakai, Tetrahedron, 2004, 60, 11783–11803 CrossRef CAS.
- H. Brockmann Jr. and J. Bode, Justus Liebigs Ann. Chem., 1974, 1017–1027 CrossRef.
- K. M. Smith and D. A. Goff, J. Am. Chem. Soc., 1985, 107, 4954–4964 CrossRef CAS.
- K. Maruyama, H. Yamada and A. Osuka, Chem. Lett., 1989, 833–836 CrossRef CAS.
- N. W. Smith and K. M. Smith, Energy Fuels, 1990, 4, 675–688 CrossRef CAS.
- K. Maruyama, H. Yamada and A. Osuka, Photochem. Photobiol., 1991, 53, 617–626 CrossRef CAS.
- R. J. Abraham, A. E. Rowan, N. W. Smith and K. M. Smith, J. Chem. Soc., Perkin Trans. 2, 1993, 1047–1059 RSC.
- H. Tamiaki, M. Amakawa, Y. Shimono, R. Tanikaga, A. R. Holzwarth and K. Schaffner, Photochem. Photobiol., 1996, 63, 92–99 CrossRef CAS.
- R. K. Pandey, S. Constantine, T. Tsuchida, G. Zheng, C. J. Medforth, M. Aoudia, A. N. Kozyrev, M. A. J. Rodgers, H. Kato, K. M. Smith and T. J. Dougherty, J. Med. Chem., 1997, 40, 2770–2779 CrossRef CAS.
- H. Tamiaki, T. Miyatake and R. Tanikaga, Tetrahedron Lett., 1997, 38, 267–270 CrossRef CAS.
- S. Mettath, M. Shibata, J. L. Alderfer, M. O. Senge, K. M. Smith, R. Rein, T. J. Dougherty and R. K. Pandey, J. Org. Chem., 1998, 63, 1646–1656 CrossRef CAS.
- H. Tamiaki, S. Yagai and T. Miyatake, Bioorg. Med. Chem., 1998, 6, 2171–2178 CrossRef CAS.
- M. Katterle, A. R. Holzwarth and A. Jesorka, Eur. J. Org. Chem., 2006, 414–422 CrossRef CAS.
- T. Miyatake, S. Tanigawa, S. Kato and H. Tamiaki, Tetrahedron Lett., 2007, 48, 2251–2254 CrossRef CAS.
- R. O. Hutchins, C. A. Milewski and B. E. Maryanoff, J. Am. Chem. Soc., 1973, 95, 3662–3668 CrossRef CAS.
- V. Nair and A. K. Sinhababu, J. Org. Chem., 1978, 43, 5013–5017 CrossRef CAS.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 3, 283–291 CrossRef CAS.
- F. Li, S. Gentemann, W. A. Kalsbeck, J. Seth, J. S. Lindsey, D. Holten and D. F. Bocian, J. Mater. Chem., 1997, 7, 1245–1262 RSC.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433–20443 CrossRef CAS.
- E. Zass, H. P. Isenring, R. Etter and A. Eschenmoser, Helv. Chim. Acta, 1980, 63, 1048–1067 CAS.
- I. D. Jones, R. C. White, E. Gibbs and C. D. Denard, J. Agric. Food Chem., 1968, 16, 80–83 CrossRef CAS.
- S. I. Yang, J. Seth, J.-P. Strachan, S. Gentemann, D. Kim, D. Holten, J. S. Lindsey and D. F. Bocian, J. Porphyrins Phthalocyanines, 1999, 3, 117–147 CrossRef CAS.
- A. T. Gradyushko, A. N. Sevchenko, K. N. Solovyov and M. P. Tsvirko, Photochem. Photobiol., 1970, 11, 387–400 CrossRef CAS.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1–13 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970, pp. 140–192 Search PubMed.
- M. Taniguchi, O. Mass, P. D. Boyle, Q. Tang, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, J. Mol. Struct., 2010, 979, 27–45 CrossRef CAS.
- M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, pp. 1–165 Search PubMed.
- M. Gouterman, J. Chem. Phys., 1959, 30, 1139–1161 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- G. Weber and F. W. J. Teale, Trans. Faraday Soc., 1957, 53, 646–655 RSC.
- Except for molecular mechanics and semi-empirical models, the calculation methods used in Spartan have been documented in Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W.-Z. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer III, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |