Gold nanoparticles decorated with a photoactivable nitric oxide donor/cyclodextrin host/guest complex
Noufal
Kandoth
,
Elisa
Vittorino
and
Salvatore
Sortino
*
Dipartimento di Scienze Chimiche, Universitá di Catania, I-95125, Catania, Italy. E-mail: ssortino@unict.it
First published on 16th November 2010
Abstract
Hybrid gold nanoclusters exhibiting photoregulated release of nitric oxide are obtained by appropriate self-assembling of a suitable host, a photoactivable guest and Au nanoparticles. The excitation of the core, the shell or both components of the nanoclusters with visible light, together with their dark stability and water solubility, make these nanoarchitectures intriguing candidates to be tested in biomedical research studies.
Noble metal nanoparticles are at the leading edge of the burgeoning field of nanomedicine.1 In particular, gold nanoparticles (AuNPs) are the subject of great interest due to their fascinating optical properties arising from the intense surface plasmon resonance in the visible spectral region. The absorption cross section of AuNPs is orders of magnitude larger than that of strongly absorbing organic molecules, allowing a large number of photons per AuNPs to be absorbed.2 This feature, along with the flexible synthetic routes and the relative biocompatibility, has made AuNPs particularly attractive in biologically related research with particular emphasis in labeling and sensing.3 In addition, AuNPs have emerged as appealing therapeutic anticancer agents due to their remarkable photothermal properties.1,3,4 In fact, a large fraction of the photon energy absorbed can be effectively converted into heat on the picosecond time regime.5 The heat dissipation from the hot AuNPs to surrounding environment can be exploited for a well recognized and promising form of noninvasive anti-cancer therapy known as hyperthermia.6 This treatment takes advantage of the higher sensitivity of tumor cells to temperature in the range 42–45 °C than normal tissue cells.7 On these grounds, the integration into the AuNPs structure of suitable units able to deliver anticancer species on demand may represent a viable strategy for fabricating intriguing nanoarchitectures with great potential in the emerging field of “bimodal” anticancer therapies. Such treatments are aimed at exploiting either additive or synergistic effects arising from the generation of two active species in the same region of space with the final goal to maximize the anticancer effects. At this regard, thiol-stabilized AuNPs, commonly referred to as monolayer protected clusters, provide a fascinating synthetic scaffold for the creation of drug delivery systems due to their versatility, superior biocompatibility and low toxicity.8
Nitric oxide (NO) is a free radical that beyond its multiple role in the regulation of key physiological processes9 has recently stimulated an upsurge of interest due to its promising anticancer activity.10 However, it should be stressed that NO can act as a double-edge sword. The biological effects of this radical have been shown to be highly site, concentration and dosage dependent, creating a complex role for the molecule in opposing beneficial and deleterious events.11 Therefore, rapidity of delivery and precise spatiotemporal control of this transient species are fundamental criteria that would be meet in view of therapeutic applications. Using light as an external trigger provides the great opportunity to satisfy both the above requisites in the case of photoactivable NO donors,12,13 with the additional advantage of not affecting important physiological parameters such as pH, temperature and ionic strength.
On the basis of these considerations, the achievement of water soluble AuNPs exhibiting photoregulated NO release is a worthwhile objective to pursue that can represent a significant step-forward in the perspective of multimodal biomedical applications. In principle, such systems may combine a dense Au core for imaging and photothermia with a photoactivable shell for controlled release of NO.
With this goal in mind, in this contribution we show that water soluble NO photoreleasing AuNPs can be achieved through a simple approach by self-assembling a host/guest complex between the thiol derivative NO photodonor 1 and α-cyclodextrin (α-CD) on the surface of colloidal AuNPs (Scheme 1).
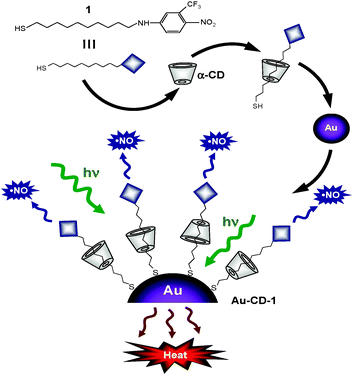 | ||
| Scheme 1 | ||
In addition to our ongoing interest in designing light-controlled NO delivering molecular assemblies,13 this work was inspired by a pioneer paper by Kaifer and co-workers reporting the surface derivatization of Au colloids through the aqueous solubilization of alkanethiols by CD derivatives.14
Thiol derivative 1 incorporates a commercial nitroaniline derivative in its molecular skeleton. We have discovered this chromogenic moiety to be a suitable NO photodonor15 as it satisfies several biological prerequisites for photocages.16 In fact it combines long term dark stability, adequate absorption coefficient at wavelengths longer than 350 nm, release of NO under the exclusive control of light inputs and generation of non-toxic and nonabsorbing photoproducts in the visible region. In particular, the NO photorelease takes place through a nitro- to nitrite photorearrangement followed by rupture of the O–NO bond with formation of NO and a phenoxy radical.17
Thiol 1 is insoluble in aqueous solution. On the other hand, it is fairly soluble in the presence of α-CD as a result of the formation of a host–guest inclusion complex. This is demonstrated by the appearance of the characteristic absorption of the nitroaniline chromophore with a maximum at ca. 400 nm (a in Fig. 1). It is generally accepted that the host complexation is primarily determined by the tightness of fit between the guest and the CD cavity. The diameter of the α-CD cavity is similar to the lateral size of the photoactive centre of 1 (ca. 0.57 nm). As a consequence, the host should preferentially accommodate a part of the hydrophobic alkyl chain of 1 most snugly (see Scheme 1), analogously to other alkanethiols with chain length comparable to 1.18 This hypothesis is corroborated by the absence of any significant induced circular dichroism signal in correspondence of the absorption bands of the nitroaniline chromophore. Circular dichroic bands were instead observed when the chromophore was encapsulated within the larger β-CD cavity.19
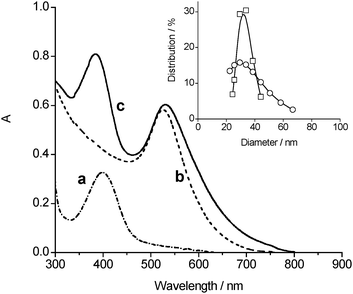 | ||
| Fig. 1 Absorption spectra of (a) 1 in the presence of aqueous 20 mM α-CD, (b) non-functionalized AuNPs and (c) Au–CD–1 in water solution. Spectrum b is reported in arbitrary absorbance, for the sake of comparison. The inset shows the particle size distribution of (□) AuNPs and (○) Au–CD–1 samples, obtained by dynamic light scattering measurements. | ||
Naked AuNPs were prepared through a typical synthetic protocol based on the reduction of AuCl4− by citrate in aqueous medium.20 They exhibit the characteristic visible plasmon absorption band at 524 nm (b in Fig. 1) and a mean diameter of 30 nm (SD 13%) (inset Fig. 1).
These AuNPs were then decorated with the 1/α-CD host/guest complex to obtain the functionalized Au–CD–1 hybrid system (Scheme 1). In a typical preparation, 1 mg of 1 was added to 50 mL of the gold colloidal water solution containing 20 mM α-CD and the resulting mixture was stirred for 3 days at room temperature while a dark-red precipitate was formed. The obtained solid was collected by centrifugation and washed several times with water first, and acetonitrile then, in order to remove the large excess of α-CD and the unreacted 1, respectively. The dried precipitate is insoluble in nonpolar solvents such as chloroform and dichloromethane but fairly soluble in water and dimethylformammide. The absorption spectrum (c in Fig. 1) clearly exhibits the absorption features of the NO photodonor together and those of the Au core. In particular, the metal plasmon absorption band is slightly shifted and broadened when compared to that of the non-functionalized AuNPs, according to the modification of the gold surface upon covalent binding of the thiol group.21 Interestingly, decoration of the AuNPs surface does not significantly alter the mean diameter since it changes from ca. 30 to ca. 33 nm. Nevertheless, the size distribution is larger (see inset Fig. 1) probably due to minor interparticle interactions. This view is also in agreement with the slight blue shift of the absorption maximum of the NO photodonor when compared with the unbound host–guest complex (spectra a and c in Fig. 1).
The solubility of Au–CD–1 in polar solvents is a first indication that the α-CD does not unthread upon chemisorption of 1, due to the presence of the bulky nitroaniline moiety and the AuNPs on the opposite sides of the alkyl chain, resulting in the achievement of a “rotaxane-like” structure with polar characteristics sketched in Scheme 1. An unambiguous confirmation of the presence of the α-CD ring in the modified AuNPs is provided by FTIR-ATR analysis carried out on freeze-dried sample of Au–CD–1 and, for comparison on α-CD (Fig. 2).
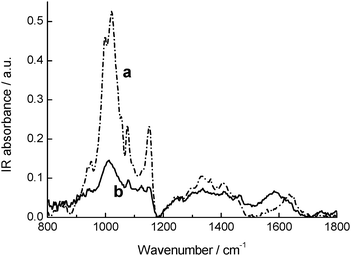
As shown in Fig. 2, the modified AuNPs show the typical C–O stretching pattern of α-CD in the range 1000–1200 cm−1. Inspection of the HOH bending band at ca. 1640 cm−1 due to crystallization water molecules of α-CD gives further important structural insights. This band shifts to lower frequency and, at the same time, increases in relative intensity in the case of Au–CD–1. This occurrence can be considered as an indication of the formation of an inclusion complex. According to recent literature data,22 the observed spectral changes are indicative of a loss of a bulk-like structural arrangement for water molecules with a corresponding establishment of H-bonded networks of increasing co-operativity. This can be ascribed to the release of intra-cavity bulk-like water molecules upon complexation, in conjunction with their substitution with less polar “guest” molecules, and their following rearrangement in interstitial H-bonded environments.
The decorated AuNPs were sensitive to visible light when irradiated in the absorption band of the nitroaniline chromophore. The typical bleaching of this band16 was in fact observed upon photolysis of a water solution of Au–CD–1 with 400 nm light, whereas the plasmon absorption band was basically unaffected (Fig. 3). The most convenient methodology to demonstrate that the observed photochemical decomposition is related to the NO release from Au–CD–1 is the direct and in real-time monitoring of this radical species. To this end, we have used an ultrasensitive NO electrode, which directly detects NO, with nM concentration resolution, by an amperometric technique. The result illustrated in the inset of Fig. 3 provides unambiguous evidence that the modified AuNPs are stable in the dark but supply NO upon illumination when irradiated with 400 nm light. The release process is strictly dependent on the external light inputs, as confirmed by the linear NO photodelivery, which promptly stops as the light is turned off, and restarts as the illumination is turned on again. Furthermore, no NO release was detected when the excitation energy was longer than 450 nm, according to the negligible absorption of the NO photodonor unit at these wavelengths.
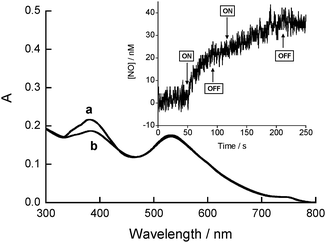 | ||
| Fig. 3 Absorption spectra of Au–CD–1 in water solution at 25 °C before (a) and after (b) 40 min of 400 nm light irradiation. The inset shows the NO release from a N2-purged water solution of Au–CD–1 upon alternate periods of light (ON) and dark (OFF) at 25 °C. | ||
Based on the absorption spectrum of Au–CD–1 reported in Fig. 1 and by taking into account that no significant change in the molar absorptivity of 1 occurs upon its binding with the metal surface (being the band profile basically unchanged), an aqueous solubility limit of ca. 50 μM (ca. 20 mg L−1) can be estimated for the Au–CD–1 nanoassembly. Given that the photoproduct generated after NO release is not absorbing at the excitation wavelength,15,16 the photochemical reaction occurs almost quantitatively, generating NO concentrations up to the micromolar range, an important requisite for anticancer applications.12,23
Photothermal effects induced by AuNPs prepared by the same protocol we used are well known in the literature.4b–e Therefore, we decided to perform some experiments in order to prove the stability of Au–CD–1 to photothermia. An aqueous sample of Au–CD–1 was at first heated for 30 min at 70 °C and then irradiated for 20 min with a 532 nm laser excitation (see Experimental details). After these treatments, the absorption spectrum remained unaffected and the changes in the nanoparticles size distribution were negligible (data not shown). Unfortunately, it was impossible to measure if NO release occurs upon the heat treatment or 532 nm light irradiation. In fact the NO detector used is very sensitive to temperature changes, resulting in false NO signals. However, as illustrated in Fig. 3, photorelease of NO is accompanied by a bleaching of the absorption band at 400 nm. The unaltered absorption spectrum we found after the above treatments provides a good experimental proof for the lack of NO generation under these stress conditions. Therefore, these findings reveal that the decorated AuNPs do not suffer neither physical nor chemical decomposition upon heating or after irradiation of the plasmon absorption band of the Au core.
In summary, we have shown the first example of AuNPs exhibiting photoregulated release of NO24 by appropriate self-assembling of three basic components. The role of the host cavity is manifold. In fact, it conveys the photoactive unit, which is insoluble in water, to the metal surface; it allows the solubilization of the resulting modified AuNPs in polar solvents and, in view of its extensive use in biocompatible materials, it is also envisaged to improve the biocompatibility of the resulting AuNPs. Au–CD–1 exhibits a combination of indispensable properties in view of bio-applications, such as small sizes, water solubility, thermal stability and photoactivation with low energy light. A further, remarkable advantage arising from the logical design of our systems is that the distinct absorption spectral region of the Au centre and the NO photodonor units allows in principle the selective excitation of the core (triggering photothermia), the shell (triggering NO release) or both (triggering bimodal effects) by the appropriate tuning of the visible light energy. These features make Au–CD–1 a multifunctional, eligible candidate for light-mediated experiments in biomedical research. In vitro studies on cancer cells addressed to evaluate the biological activity of the photoactive nanoassembly and efforts to extend its absorption spectrum to lower energy will be object of forthcoming investigation.
Experimental details
Syntheses
Compound 1 was synthesized according to our previously reported procedure.16a Naked AuNPs were prepared through reduction of AuCl4− by citrate in aqueous medium.20 These AuNPs were filtered with a 0.8 μM membrane filter, before assembling with 1/α-CD. The absence of α-CD and free 1 after washing the precipitate was verified by TLC and spectrophotometric analysis.Instrumentation
Absorption spectra were recorded with a Jasco V 560 spectrophotometer.Nanoparticle sizes were measured by a dynamic light scattering performed with a Horiba LS 550 apparatus equipped with a diode laser with a wavelength of 650 nm. Diameters are the result of averaged measurements over a time of 2 min.
FTIR-ATR spectra were recorded at room temperature using a Bomem DA8 Fourier transformer Spectrometer, operating with a Globar Source in combination with a KBr beamsplitter and a DTGS/KBr detector. The powders were contained in a Golden Gate diamond ATR system, based on the attenuated total reflectance (ATR) technique and covering a wavelength range from 600 to 4000 cm−1. In this configuration the evanescent wave will be attenuated in region of the IR spectrum where the sample absorbs energy. The spectra were recorded with a resolution of 2 cm−1, averaging 100 scans to achieve a good signal-to-noise ratio and highly reproducible spectra. No smoothing was applied and spectroscopic manipulation such as baseline adjustment and normalization was performed using the Spectracalc software package GRAMS (Galactic Industries).
Photochemical experiments
Steady-state irradiation was performed in a thermostated quartz cell (1 cm pathlength, 3 ml capacity) by using a Rayonet photochemical reactor equipped with 8 RPR lamps with an emission in the 380–480 nm range with a maximum at 420 nm in the presence of a 400 nm cut-off filter. Pulsed laser irradiation was performed by means of the second harmonic of a Nd-YAG Continuum Surelite II-10 laser system (λexc = 532 nm, pulse width 6 ns FWHM, E532 ≈ 20 mJ per pulse).The NO photorelease experiments were performed by irradiating the samples in the same cell as above using the monochromatic radiation (λexc = 400 nm) of a fluorimeter Fluorolog-2 (mod. F-111), under continuous stirring.
NO detection
NO release was measured with a World Precision Instrument, ISO-NO 1000 meter, equipped with a data acquisition system, and based on direct amperometric detection of NO with short response time (<5 s) and sensitivity range 1 nM–20 μM. The analog signal was digitalized with a four-channel recording system and transferred to a PC. The sensor was accurately calibrated by mixing standard solutions of NaNO2 with 0.1 M H2SO4 and 0.1 M KI according to the reaction:| 4H+ + 2I− + 2NO2− → 2H2O + 2NO + I2 |
Acknowledgements
We thank the Marie Curie program #237962 CYCLON (FP7-PEOPLE-ITN-2008) for financial support to MC fellow, N.K. and funding of the research. Financial support from MIUR (PRIN 2008), Rome, Italy is also acknowledged. We finally express our sincere thanks to Dr Valentina Venuti (Università di Messina) for her inestimable help in the FTIR-ATR measurements and related discussion, and to the anonymous referees for their constructive comments.Notes and references
- See, for example: P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 Search PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410 CrossRef CAS.
- See, for example: (a) R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896 RSC; (b) M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084 RSC.
- (a) X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Lasers Med. Sci., 2008, 23, 217 CrossRef; (b) I. H. El-Sayed, X. Huang and M. A. El-Sayed, Cancer Lett., 2006, 239, 129 CrossRef CAS; (c) A. Mazzaglia, M. Trapani, V. Villari, N. Micali, F. Marini Merlo, D. Zaccaria, M. T. Sciortino, F. Previti, S. Patanè and L. Monsù Scolaro, J. Phys. Chem. C, 2008, 112, 6764 CrossRef CAS; (d) Y. Haba, C. Kojima, A. Harada, T. Ura, H. Horinaka and K. Kono, Langmuir, 2007, 23, 5243 CrossRef CAS; (e) C. D. Jones and L. A. Lyon, J. Am. Chem. Soc., 2003, 125, 460 CrossRef CAS.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409 CrossRef CAS.
- A. O. Govorov, W. Zhang, T. Skeini, H. Richardson, J. Lee and N. A. Kotov, Nanoscale Res. Lett., 2006, 1, 84 CrossRef.
- (a) M. R. Choi, K. J. Stanton-Maxey, J. K. Stanley, C. S. Levin, R. Bardhan, D. Akin, S. Badve, J. Sturgis, J. P. Robinson, R. Bashir, N. J. Alas and S. E. Clare, Nano Lett., 2007, 7, 3759 CrossRef CAS; (b) J. Chen, B. Wiley, Z. Y. Li, D. Campbell, F. Saeki, H. Cang, L. Au, J. Lee, X. Li and Y. Xia, Adv. Mater., 2005, 17, 2255 CrossRef CAS.
- (a) M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225 CrossRef CAS; (b) S. S. Agasti, A. Chompoosor, C.-C. You, P. Ghosh, C. K. Kim and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 5728 CrossRef CAS; (c) R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289 CrossRef CAS.
- (a) Nitric Oxide: Biology and Pathobiology, ed. L. J. Ignarro, Elsevier Inc., 2010 Search PubMed; (b) L. J. Ignarro (ed), Special Journal Issue on Nitric Oxide Chemistry and Biology, Arch. Pharmacal Res., 2009, 32.
- D. Fukumura, S. Kashiwagi and R. K. Jain, Nat. Rev. Cancer, 2006, 6, 521 CrossRef.
- Nitric Oxide Donors for Pharmaceutical and Biological Applications., ed. P. G. Wang, T. B. Cai and N. Taniguchi, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005 Search PubMed.
- A. D. Ostrowski and P. C. Ford, Dalton Trans., 2009, 10660 RSC , and references therein.
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903 RSC , and references therein.
- J. Liu, R. Xu and A. E. Kaifer, Langmuir, 1998, 14, 7337 CrossRef CAS.
- S. Conoci, S. Petralia and S. Sortino, U.S. Pat. Appl. Publ., Appl. No. PCT/IT2006/000575, 2009, pp. 20 Search PubMed.
- (a) E. B. Caruso, S. Petralia, S. Conoci, S. Giuffrida and S. Sortino, J. Am. Chem. Soc., 2007, 129, 480 CrossRef CAS; (b) M. Barone, M. T. Sciortino, D. Zaccaria, A. Mazzaglia and S. Sortino, J. Mater. Chem., 2008, 18, 5531 RSC.
- Phenoxyl radicals are known to be scarcely reactive towards biological targets. (a) G. E. Burton and K. U. Ingold, Acc. Chem. Res., 1986, 19, 194 CrossRef CAS.
- (a) J. Yan and S. Dong, J. Electroanal. Chem., 1997, 440, 229 CrossRef CAS; (b) J. Yan and S. Dong, Langmuir, 1997, 13, 3251 CrossRef CAS; (c) S. Mittler-Neher, J. Spinke, M. Liley, G. Nelles, M. Wessei, R. Back, G. Wenz and W. Knoll, Biosens. Bioelectron., 1995, 10, 903 CrossRef CAS.
- S. Sortino, S. Giuffrida, G. De Guidi, R. Chillemi, S. Petralia, G. Marconi, G. Condorelli and S. Sciuto, Photochem. Photobiol., 2001, 73, 6 CrossRef CAS.
- H. S. Zhou, S. Aoki, I. Honma, M. Hirasawa, T. Nagamune and H. Komiyama, Chem. Commun., 1997, 605 RSC.
- J. Zhang, J. K. Whitesell and M. A. Fox, Chem. Mater., 2001, 13, 2323 CrossRef CAS.
- (a) F. Mallamace, M. Broccio, C. Corsaro, A. Faraone, D. Majolino, V. Venuti, L. Liu, C.-Y. Mou and S.-H. Chen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 424 CrossRef CAS; (b) V. Crupi, D. Majolino, F. Longo, P. Migliardo and V. Venuti, Vib. Spectrosc., 2006, 42, 375 CrossRef CAS.
- E. V. Stevens, A. W. Carpenter, J. H. Shin, J. Liu, C. J. Der and M. H. Schoenfisch, Mol. Pharmaceutics, 2010, 7, 775 CrossRef CAS.
- Synthesis of Au nanoparticles releasing NO in the dark has been reported in an excellent paper by Schoenfisch and coworkers. (a) A. R. Rothrock, R. L. Donkers and M. H. Schoenfisch, J. Am. Chem. Soc., 2005, 127, 9362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |