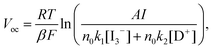DOI:
10.1039/C0NJ00653J
(Paper)
New J. Chem., 2011,
35, 127-136
Aryl/hetero-arylethyne bridged dyes: the effect of planar π-bridge on the performance of dye-sensitized solar cells†
Received
(in Montpellier, France)
21st August 2010
, Accepted 15th September 2010
First published on 18th October 2010
Abstract
We have designed and synthesized three metal-free sensitizers consisting of dimethoxy-substituted triphenylamine as donor, cyanoacrylic acid as acceptor with planar π-bridge. The photon to current conversion efficiency of these dyes were measured as sensitizers for DSSCs analysis. The results obtained from these experiments show that the overall conversion efficiency of D3 gave the highest overall efficiency 4.98% with short circuit current density (Jsc) of 11.29 mA cm−2, an open-circuit voltage (Voc) of 0.637 V and a fill factor of 69.2% under standard global AM 1.5 solar conditions. In addition, the experimental results show that π-conjugated bridge can extend the absorption of dye molecular and strong planarity (close-packing) can suppress the charge recombination at TiO2/electrolyte interface. Hence, the donor-π bridge-acceptor architectures can generate a unidirectional current flow, to some extent, which can enhance the cell performance.
Introduction
Even though silicon and other semiconductor-based solar cells have dominated the solar-cell market for decades, dye-sensitized solar cells (DSSCs) have attracted much consideration due to their low cost and relatively high power conversion efficiency (η) in the past few years.1–3 In DSSCs systems, the dye sensitizers play an important role and many research groups have devoted to exploring novel ruthenium (Ru) polypyridyl complexes, such as cis-dithiocyanate-N,N′-bis(4-carboxylate-4-tetrabutyl-ammonium-carboxylate-2,2′-bipyridine)-Ru(II) (N719) that could improve the cell performance and push the efficiency above 10% under air mass 1.5 global (AM 1.5G) irradiation.4–6 Promoted by continuous material innovation and the systematic study of device fabrication in conjugation with highly volatile electrolytes, the cells with the ruthenium polypyridyl sensitizers have reached high efficiencies of 11.1% measured under the AM 1.5G conditions.7 Due to the limited ruthenium resources and the heavy-metal toxicity, metal-free organic dyes have received vital research interest. In general, the metal-free sensitizers for DSSCs mainly comprise a large conjugated system (donor-π bridge-acceptor moieties) which can expand the absorption spectrum and thereby increase the short circuit current (Jsc).6 Furthermore, the rate of charge transfer within molecules can also be enhanced by intramolecular donor–acceptor delivery.8 However, introducing more π-conjugated bridge moiety to conjugated dyes will lead to unfavorable π-stacked dye aggregation on TiO2, which may result in a decrease in DSSCs performance.9 Consequently, an appropriate bridge system is of the essence to obtain the high power conversion efficiency of organic dye sensitized solar cells. In this regard, these two factors should be adjusted to reach a compromise when designing novel dyes to improve the overall efficiency of DSSCs. Recently, several metal-free organic dyes including coumarin,10–12merocyanine,13–15indoline,16,17polyene,18hemicyanine,19,20triphenylamine,21–23fluorine24–26 and tetrahydroquinoline27 as the donating group have been developed for DSSCs. Thus far, the highest photoelectric conversion efficiency of metal-free dye sensitized solar cells was reported to be around 9 to 9.8%.28 It is well accepted that 4-methoxy-triphenylamine moiety as a donor in metal-free sensitizer can effectively suppress the dye-aggregation.29–31 On the other hand, cyanoacrylic acid as an acceptor is better than other anchoring groups such as rhodanine-3-acetic acid due to its coplanarity with respect to spacer unit and good electron coupling with TiO2.22,33–34 In addition to the intrinsic properties of donor and acceptor, the electron transfer can also be improved when the bridge moiety can be turned on or off on demand in donor–bridge–acceptor (D–B–A) system. According to Geppert and Vivie-Riedle's results,35 the bridge molecule can suppress the electron transfer when the molecule is in the closed form, while in the open form, the molecule can allow the electron transfer from D via B to A. Moreover, experimental results indicated that linear bridge moiety with good planarity can supress the electron recombination.36
In view of the key role of bridge moiety in DSSCs systems, triphenylamine moiety has been extensively used in DSSCs as donor units due to slowing down the recombination reactions in the cell,30–32 Here, we reported the design and synthesis of three new organic dyes bearing suitable linear π-conjugated bridge moieties with good planarity as metal-free sensitizers for DSSCs (D1, D2 and D3 as shown in Scheme 1). The electrochemical and photovoltaic properties of these metal-free dyes have also been investigated.
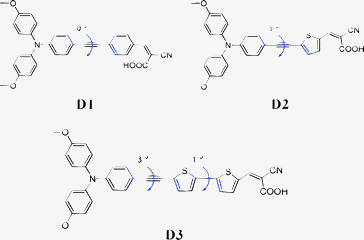 |
| | Scheme 1 Molecular structures of dyes (D1–D3). | |
Results and discussion
Synthesis
The three new metal-free organic dyes are made of three important parts such as donor group (dimethoxy-substituted triphenylamine system), a conjugated spacer (alkyne bridged systems) and acceptor group (cyanoacrylic acid). The following three important points made us to design the arylethyne bridged metal-free organic dyes, namely (1) the donor group such as triphenylamine may effectively inhibit I3− by making denser film formation on surface of the TiO2 film, (2) the aryl/heteroarylethyne spacer is a semirigid conjugated linker37 which should support electron injection and (3) the cyanoacrylic acid group acts as acceptor group and also anchoring group.
The synthetic outlines of the new metal-free organic dyes are shown in Schemes 2–5. The essential intermediates were achieved according to literature methods.14,38,39 The dimethoxy-substituted triphenylamine donor group was synthesized by Buchwald coupling and the yield was only 30% (Scheme 2). On compared to the previous report,40 the synthetic strategy employed for arylethyne based dyes is quite facile and the yield of these dyes obtained by this synthetic route is also moderate. It involves only two important steps to attain these organic dyes such as (1) Sonogashira cross-coupling of the ethynyl unit with halogen unit to afford the corresponding aldeheydes as precursors, and (2) Knoevenagel condensation of the precursor aldehydes with cyanoacetic acid afforded the corresponding metal-free organic dyes. The precursor aldehydes 7 and 11 were achieved by Sonogashira coupling method using dichlorobis(triphenylphosphine) palladium(II) mediation afforded the yield of 51% and 50% respectively (Scheme 3 and 4).
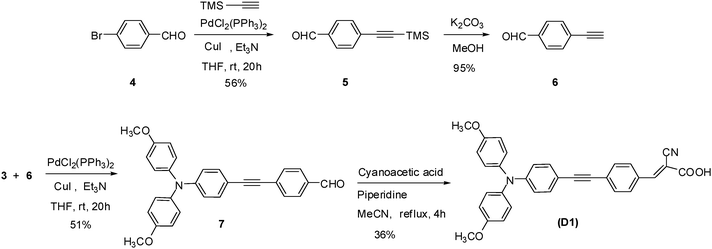 |
| | Scheme 3 Synthetic route of the D1 sensitizer. | |
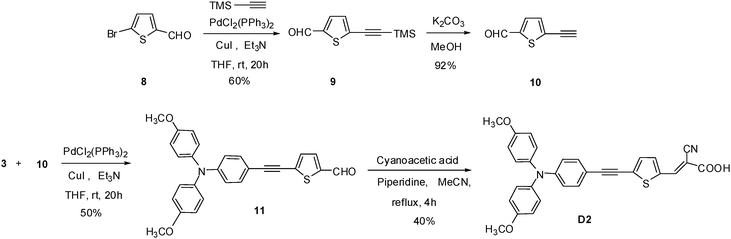 |
| | Scheme 4 Synthetic route of the D2 sensitizer. | |
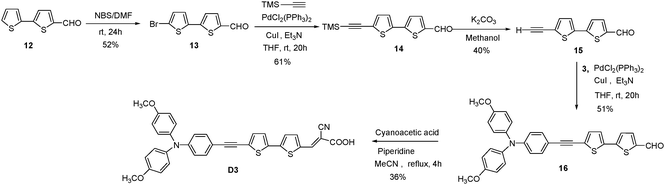 |
| | Scheme 5 Synthetic route of the D3 sensitizer. | |
These intermediates were converted into corresponding dyes by treating with cyanoacetic acid using piperdine as base in a yield of 36% (D1) and 40% (D2), respectively. The dye D3 was obtained from building unit 14 by Sonogashira followed by Knoevenagel condensation41 in a yield of 36% (Scheme 5). The chemical structures of the new four metal-free organic dyes were characterized by 1H and 13C NMR spectroscopy, EI mass spectroscopy, and high-resolution mass spectroscopy.
Absorption properties in solution and on TiO2 film
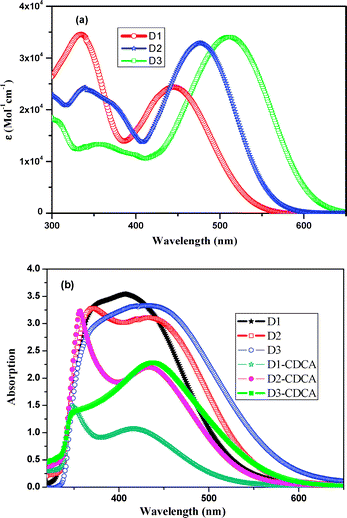
The UV-visible absorption spectra of the three metal-free organic dyes in CHCl3 solution and on TiO2 films are shown in Fig. 1. The molar extinction coefficients decrease in the order of D1 > D2 > D3 in ultraviolet region, while opposite order is observed in the visible region and the spectra exhibit corresponding red-shift. The bands at around 350 nm are attributed to the π–π* transition of the conjugated molecule and intramolecular transfer between the donor and the acceptor.29 The positions of the bands are strongly influenced by the extended conjugation and bathochrome group. The hyperchromic effect intensity of bands in UV region might be attributed to introducing planar macrocyclic structure to the bridge moiety, while the intensity of bands in visible region can be enhanced by introducing auxochrome group. The π-conjugation groups would facilitate the absorption spectra by bathochromic shift towards near IR region, which causes an improved match of the absorption bands of sensitizer to the solar radiation. It is obvious that the absorption bands of thiophene are at a much longer wavelength range due to substantial resonance enhancement effect compared with benzenoid moieties.42,43 The calculated torsion angles with respect to the conjugated spacers in three D–B–A dyes are shown in Scheme 1. D1 has a negligible torsion angle compared with D2 and D3, and all the three dyes have small torsion angle. This indicates that triple bond can enhance the planarity compared with the other similar dyes as reported.29,40,43,44c Compared with the spectra in CHCl3 solution, the absorption bands of these dyes on TiO2 films exhibit larger red-shift and significant spectral broadening due to the interaction of the anchoring groups with the surface of TiO2 and the J-aggregation of dye molecules.44 At the same time, the absorption bands are broadened due to the aggregation of dyes on TiO2. To investigate the degree of aggregation for these three dyes, we dipped TiO2 film into dye solution including 0.5 mM dye and 10 mM chenodeoxycholic acid (CDCA) for 12 h. It was found that the CDCA co-adsorbent can prevent the aggregation on TiO2 film, which is consistent with those previously reported.44d
Compared with D1 and D3, the aggregation of D2 onto the TiO2 surface dimished in 10 mM CDCA in sensitizer solution. The effect of CDCA as the coadsorbent on cell performance needs to be investigated in details. The emission spectra of these dyes were measured in CHCl3 (Fig. S1, ESI†). The first emission peaks of D1 (598 nm) and D2 (614 nm) are hypso-chromically shifted by 50–60 nm relative to D3 (661 nm), which is consistent with the results of the UV-Vis absorption spectra in CHCl3.
Electrochemical properties
Cyclic voltammetry was employed to determine the oxidation potentials of the three metal-free organic dyes. The measurements were performed in CHCl3 solution with 0.2 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) using ferrocene as an internal standard at 0.63 V vs.NHE (Fig. S2, ESI†). The oxidation potential and the reduction potential correspond to the HOMO and LUMO energies, respectively. The oxidation potentials of D1, D2 and D3 are 1.00 V, 0.99 V and 1.00 V vs.NHE, respectively, which are more positive than the that of I−/I3− couple (0.5 V vs.NHE),44 ensuring the regeneration of sensitizer radical cations by the electrolyte redox couple. By neglecting any entropy change during the electron-injection process,32–34 the excited-state oxidation potential (E(S+/S*)) of the sensitiser can be derived from the ground-state oxidation couple (E(S+/S)) and the 0–0 excitation energy (E(0–0)) according to eqn (1)| | | E(S+/S*) = E(S+/S) − E(0–0) | (1) |
Transition energy E(0–0) was estimated from the interception of the absorption and emission spectra in CHCl3 solution. As shown in Table 1, E(0–0) energies for D1, D2 and D3 were extracted to be 2.37 eV, 2.28 eV and 2.18 eV, respectively. The calculated excited-state oxidation potentials for D1, D2 and D3 are −1.37 V, −1.29 V and −1.18 V vs.NHE, respectively, which are more negative than the equivalent potential for N719 sensitizer (−0.98 V vs.NHE) and the conduction band (CB) of TiO2 (−0.5 V vs.NHE). Therefore, there are sufficient driving forces for charge injection to the conduction band of semiconductors.46 From spectroscopic and electrochemical measurements, driving forces, ΔGinj, for electron injection from the dye excited singlet state to the CB of TiO2 (−0.5 V vs.NHE), and ΔGreg, the regeneration of the dye radical cation by electrolyte redox couple (+0.5 V vs.NHE) for the dye-sensitized solar cells are extracted and shown in Table 1. From these data, it can be seen that both of the processes are thermodynamically feasible.
|
Dye
|
Absmaxa/nm |
ε/M−1 cm−1 |
E
ox
b/V vs.NHE |
E
0–0
c/V |
E
ox*d/V vs.NHE |
ΔGinje/eV |
ΔGregf/eV |
|
Absorption and emission spectra of D1, D2 and D3 in chloroform solution.
Measured ground-state oxidation potential of the dyes. Potentials measured vs. Fc+/Fc were converted to normal hydrogen electrode (NHE) by addition of +0.63 V.
0–0 transition energy, E(0–0), estimated from the intercept of the normalized absorption and emission spectra in chloroform.
Estimated LUMO energies, E(LUMO)vs.NHE from the estimated highest occupied molecular orbital (HOMO) energies obtained from the ground-state oxidation potential by adding the 0–0 transition energy, E(0–0).
Driving forces for electron injection from the dye excited singlet state (Eox*) to the conduction band of TiO2 (−0.5 V vs.NHE).
Driving forces for the regeneration of the dye radical (Eox) by I−/I3−redox couple (+0.5 V vs.NHE).
|
|
D1
|
446 |
24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 402 402 |
1.00 |
2.37 |
−1.37 |
−0.87 |
−0.50 |
|
D2
|
476 |
33053 |
0.99 |
2.28 |
−1.29 |
−0.79 |
−0.49 |
|
D3
|
511 |
34604 |
1.00 |
2.18 |
−1.18 |
−0.68 |
−0.50 |
To gain insight into the electronic structures of excited states, we performed time-dependent density functional theory (TD-DFT) calculations on these three organic dyes. We calculated the contribution of singly excited state configurations to each electronic transitions and the molecular orbital contributions to analyze the charge-separated states of these dyes. All the three dyes have small torsion angle as shown in Scheme 1. The calculated molecular orbital energy diagrams and isodensity surface plots for three dyes D1, D2 and D3 are shown in Fig. 2. The HOMO–LUMO band gap of D1, D2 and D3 three dyes is 2.18 eV, 2.17 eV and 2.01 eV, respectively. D3 has the smallest bandgap, causing the red-shifted absorption spectra, which is ideally preferred for DSSC application. Compared with D2 and D3 with two thiophene units, it has lower a LUMO–HOMO gap due to the increased conjugation along the dye, which is consistent with the previous reports. Meanwhile, the LUMO of D3 is closer to the conduction band of TiO2 than that of D1 and D2. This result shows that the extended π-system could promote the intramolecular charge transfer and charge separation behavior from bridge group to acceptor group after photo-excitation.3b,c Compared with D2, the HOMO level of D3 (vs.NHE) is more positive, indicating that the regeneration of the oxidized dyes with I− ions is thermodynamically possible.1c Based on the calculated molecular orbital contribution (Table S1, ESI†), we found that, among three dyes, D3 exhibits the most obvious intramolecular charge seperation. This means that D3 possesses more stronger photo-induced intramolecular charge transfer properties capability than D1 and D2, and the major portions of the D3 HOMO are located on the donor moiety. LUMO contains the acceptor moiety, the triple bond and the bridge, which strongly suggests that electrons move from donor moiety, passing through bridge moiety, to the acceptor and form linearly photoexcitation-induced intramolecular charge transfer. When the π-conjugation increased, the overlapping extension can be enhanced by the bridge which might improve the electronic coupling between the dye and TiO2 film. The excitation energies of D1, D2 and D3 were also compared by TD-DFT calculation results and shown in Table S2 (ESI†). The results clearly indicate that introduction of thiophene group in the bridge moiety increases the oscillator strength of the visible range, which could give rise to the enhancement of the solar spectrum overlap. In contrast, introducing phenyl group in the bridge moiety can obviously increase the oscillator strength in ultraviolet region though phenyl group which makes against the bathochromic effect. Both the two dominating excited states of these three dyes originated from HOMO → LUMO and HOMO-1 → LUMO contributions and the intensities are coincident with the experimental absorptions of these three dyes. Thus, the transitions give rise to an intramolecular charge flow from the donor moiety to the acceptor moiety. Potentially, the resulting excited state is strongly coupled to the semiconductor surface, due to charge delocalization involving the cyanoacetic acid anchoring group.29–31,35,42–44
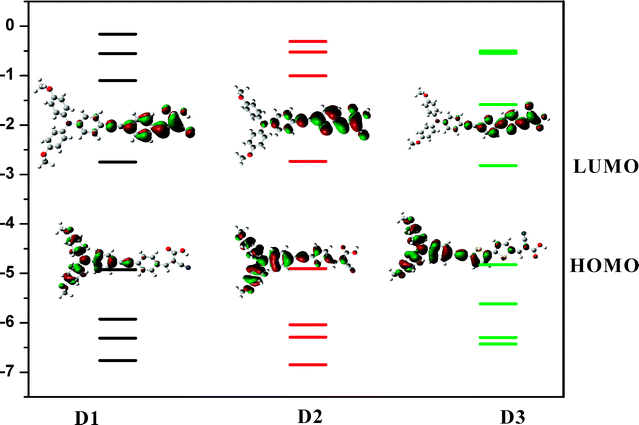 |
| | Fig. 2 Molecular orbital energy diagrams (TD-B3LYP/6-31G*//B3LYP/6-31G* in chloroform using C-PCM framework) and isodensity surface. | |
The incident monochromatic photon-to-current conversion efficiency (IPCE) values for the three organic dyes are shown in Fig. 3. The IPCEs are represented by eqn (2):
| | 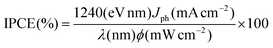 | (2) |
where
Jph is the short-circuit photocurrent density for monochromatic irradiation,
λ and
ϕ are the wavelength and the intensity of the monochromatic light, respectively, The highest IPCE values for
D1,
D2 and
D3 are found to be around 80%, 80% and 90%, respectively. There are some red-shift in the IPCE compared to the absorption spectra in
Fig. 1, which can be attributed to the aggregation of the
dyes on TiO
247–49 and the interaction of the anchoring groups with the TiO
2 surface.
30,43 These results also show that the absorption intensity in 350–400 nm region can be increased by the rigid functional group because of the high molar extinction coefficient as well as p-π-conjugated bridge which would extend the absorption of
dye molecule as mentioned above.
 |
| | Fig. 3
IPCE values for DSSCs based on the three organic dyes. | |
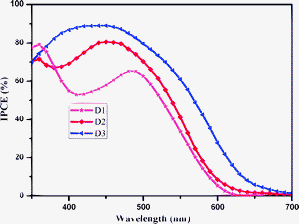
The photocurrent–voltage curves of the sensitizers are shown in Fig. 4. In this work, we selected Z907dye as control in comparison with our three organic dyes and all devices were fabricated under similar conditions. The short-circuit current densities, Jsc, open-circuit voltages, Voc, fill factors, ff, and power conversion efficiencies of the cells, η, are summarized in Table 2. The D3-sensitized cell device gives the best performance and offers Jsc of 11.39 mA cm−2, Voc of 0.637 V and ff of 69.2%, respectively. The overall conversion efficiency η can be calculated according to eqn (3), where Pin represents the intensity of the incident light (mW cm−2). Among the three organic dyes, D3 shows the highest efficiency of 4.98% which reaches around 82% of the value for Z907 under similar conditions. D2 shows an efficiency of 3.86%, while D1 gives the lowest overall efficiency of 3.21%. The poor efficiency of D1-sensitized cell is attributed to the lower light-harvesting capability in the visible range, evidenced by the UV-vis absorption spectra (Fig. 1), while D3 shows the hightest Jsc, which is consistent with its broad absorption spectra. The open circuit bias (Voc) increase in the order of D1 < D3 < D2.
| | | η = Jsc × Voc × ff/Pin | (3) |
 |
| | Fig. 4 Photocurrent density–photovoltage curve of three dyes sensitized cells under AM 1.5 G radiation (100 mW cm−2). | |
Table 2
DSSC performance parameters of various dyesa
|
Dye
|
J
sc/mA cm−2 |
V
oc/V |
ff (%) |
η (%) |
|
Performance of DSSC was measured with a working area of 0.16 cm2.
|
|
D1
|
6.95 |
0.623 |
74.2 |
3.21 |
|
D2
|
8.04 |
0.684 |
70.1 |
3.86 |
|
D3
|
11.29 |
0.637 |
69.2 |
4.98 |
|
Z907
|
15.11 |
0.686 |
58.9 |
6.11 |
Under illumination condition, VOC of the cell is potential difference between the quasi-Fermi level of the electrons (EFn*) in the nanocrystalline TiO2 film and Eredox of the electrolyte, also VOC depends logarithmically on the inverse concentration of I3− and increases with incident photo flux according to the following equation,44a
where
k1,
k2 are the kinetic constants for the back reaction of injected electrons with triiodide (I
3−) and recombination of these electrons with oxidized
dye (D
+), respectively,
RT/
βF is the thermal voltage and
n0 is the concentration of accessible electronic states in the conduction band. Since the recombination of
dye can be omitted, the electron recombination between the injected electron from the conduct band of TiO
2 and the triiodide could reduce the
Voc.
43b According to the similar results reported previously, the aggregation of the
dye and the orientation of the
dye on TiO
2 surface can aggravate charge recombination. The rigid conjugate molecules are easy to aggregate due to strong dipole–dipole interaction.
44b–d What's more, when the
dye molecules are adsorbed on TiO
2 surface, which can affect the fermi level of conduction band of TiO
2, resulting in a negative shift and this shift is increased with the increasing dipole at the
dye/TiO
2 interface.
44e This band-edge shift is more significant than the increase in recombination rate. Thus, the
Voc of
D2 gave the highest value and the
D1 gave the lowest value and due to the different level of π-stacked aggregation and band-edge shift.
44–45
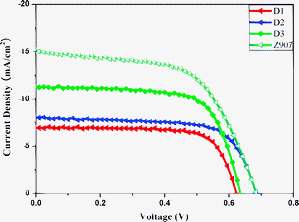
To further investigate the internal resistance, the kinetics of charge transfer and recombination in the devices, we carried out the electrochemical impedance spectroscopy (EIS) measure-ments in the dark and under illumination conditions. Fig. 5 shows the Nyquist plots for the devices in the dark and under illumination conditions. The equivalent circuit is shown in Scheme 6. Rs represents the series resistance consisting of all ohmic components in the device, R1 and R2 are charge transfer resistance components forming a parallel circuit with constant phase elements (CPE1 and CPE2) and W1 is the Finite-Length Warburg element for Nernst diffusion impedance. Three semicircles were observed at various frequency range. The first semicircle in the high-frequency region is attributed to the redox reaction of I−/I3− at the Pt/electrolyte interface, the central semicircle denotes the electron transfer at the oxide/dye/electrolyte interface and the low frequency one is caused by ion diffusion within the electrolyte.47–49 the dark condition, the impedance spectra were obtained under forward bias (−0.65 V), the charge-transfer resistance related to the recombination of electron, Rk, was estimated from the diameter of the central circle. The Rk value increases in the order of D3 (69.6 Ω) < D2 (129.3 Ω) < D1 (175.5 Ω). In the dark condition, under reverse bias, TiO2 is an insulator and the dye molecules with different dielectric constant, which are also insulating in the ground state and hence the injected electrons through TiO2/dye were coupled to that of I−/I3− ions in electrolyte. Then the dye coating as tightly packed insulating monolayer leads to a barrier blocking the dark current,44c,45 and as mentioned above, the bridge molecule can suppress the electron transfer when the molecule is in the closed form;36 accordingly, the recombination resistance at the TiO2/dye/electrolyte interface decreased according to this order of D3 < D2 < D1 may be result from those factors. Under illumination (50 mW cm−2, open-circuit voltage (OCV)) conditions, which gives rise to a new processes at the TiO2/dye/electrolyte interface including the electron injection from excited dye to TiO2, recombination with the oxidised dye, and regeneration of the sensitizer.44a The diameter of the intermediate-frequency semicircle in the Nyquist plot increases in the order of D3 < D2 < D1, which indicates that the electrons generation and transport are enhanced in the order of D3 > D2 > D1. From these data, it can be seen that planar conjugation bridge can retard the charge recombination, large conjugated bridge can increases the intramolecular charge transfer and the charge-injection rate. The cell performance can be improved by harmonizing these factors.
 |
| | Fig. 5
Nyquist plots of three films in the dark (inset) and illumination condition. | |
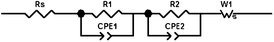 |
| | Scheme 6 Equivalent circuit of used to fit Nyquist plots. | |
Conclusions
In summary, we have designed three new linear dye molecules with good planarity and investigated the effect of the bridge moiety on the properties of dye-sensitized solar cells. The results based on UV-Vis absorptions, TD-DFT calculations, photovoltaic experiment and EIS show that π-conjugated bridge can extend the absorption of dye molecule and strong planarity (closed-packing) can suppress the charge recombination at TiO2/electrolyte interface. Improving the cell performance has been demonstrated for the dye molecules with large-conjugated planarity and p-π conjugation as bridge due to the retarded recombination and enhanced light harvesting, which provides a skilful strategy to explore metal-free sensitizers with high photo-to-current conversion efficiency. To further improve the cell performance of DSSCs based on metal-free dyes, we will focus on the development of novel organic sensitizers with large planar donor-π bridge-acceptors in the future. Currently, the study of the coadsorbent effect on these conjugated molecules and the rate of charge transfer between the sensitizers and TiO2 surface is in progress.
Experimental details
Materials and instrumentation
All reactions were carried out under Ar atmosphere. Solvents were dried by standard procedures. cis-Bis(isothiocyanato)(2,2′-bipyridyl-4,4′-dicarboxylato)(2,2′-bipyridyl-4,4′-di-nonyl)ruthenium(II) (known as Z-907, dyesol) and electrolyte (EL-HPE) were obtained from Dyesol Company. 1H and 13C NMR spectra were recorded on JEOL 400 MHz Instrument. Mass spectra under the conditions of electron impact (EI) ionisation were recorded on Thermo Finnigan MAT 95XP spectrometer. Absorption spectra were measured on JASCO V-670 UV-Vis-Near IR spectrophotometer and photoluminescence analyses were performed on a Cary Eclipses fluorescence spectrometer. Cyclic voltammetric (CV) experiments were conducted with a computer-controlled Eco Chemie Autolab PGSTAT 100 with an ADC fast scan generator. Working electrodes were 1 mm diameter planar Pt disks, used in conjunction with a Pt auxiliary electrode and an Ag/Ag+ reference electrode connected to the test solution via a salt bridge containing 0.2 M tetrabutylammonium hexafluoro-phosphate (Bu4NPF6) in CHCl3. Accurate potentials were obtained using ferrocene as an internal standard.
Calculation method
We performed time-dependent density functional theory TD-DFT calculation by using the Gaussian 03 program package.50 The geometries of the three metal-free organic dyesin vacuo were optimized using density functional theory (DFT) with the B3LYP density functional and the 6-31G* basis set.50 All optimized geometries were subjected to vibrational frequency analysis, and characterized as a minimum (no imaginary frequencies). The time-dependent DFT (TD-DFT) at the level of B3LYP/6-31G* was carried out to study the excited state of selected molecules. The absorption spectra were simulated by the 20 lowest spin-allowed singlet transitions and the C-PCM frameworks were used to describe the electrostatic solute–solvent interactions.50 The contribution of single excited state configurations to each electronic transition of the three metal-free organic dye analogues were calculated using the Swizard program, version 4.2.51 Graphical molecular orbital were generated with the Gauss View software program (version 3.09)50 and the molecular orbital contributions from the groups of atoms were obtained from the VMOdes A 7.1.51
Electrochemical impedance measurement
Electrochemical impedance experiments were carried out in the dark and under illumination condition using an Autolab electrochemical workstation with a frequency range from 0.1 Hz to 1 MHz and a potential modulation of 10 mV. The forward bias is −0.65 V and open-circuit voltage in the dark and under illumination condition, respectively. The results were fitted with the Z-view software (ZView 3.1c, Scribner Associate Inc.) to appropriate equivalent circuits. The electrochemical measurements were done using a 20 W halogen lamp as light source.
Photovoltaic characterization
The current–voltage measurement of DSSCs were performed under one sun condition (air mass (AM) 1.5) using a Sun 2000 solar simulator light source (Abet-technologies, USA) and the incident light intensity was calibrated using a radiant power meter (Oriel, 70260). The current–voltage characteristics of the cell under these conditions were obtained by applying external potential bias to the cell and measuring the generated photocurrent with a Keithley model 2440 digital source meter (Keithley, USA). The incident photon to collected electron conversion efficiency (IPCE) measurement was collected using Oriel QE/IPCE Kit equipment.
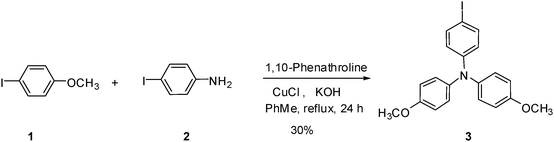
Synthesis
N-(4-Iodophenyl)-4-methoxy-N-(4-methoxyphenyl)benzene amine (3).
4-Iodoanisole (5 g, 21.36 mmol), 4-iodoaniline (2 g, 9.13 mmol) and 1,10-phenanthroline (0.30 g, 1.70 mmol) were dissolved in toluene (50 mL). After the solution was heated to 100 °C, CuCl (0.16 g, 1.70 mmol) and KOH (3.83 g, 68.29 mmol) were added under Ar. The mixture was refluxed for 12 h. After cooling to room temperature, acetic acid (5 mL) and toluene (20 mL) were added. The mixture was washed with H2O (100 mL) three times and the organic phase was dried over Na2SO4. After removal of the solvent, the residual was purified on a silica gel column (ethyl acetate–hexane: 1/9) to afford the target compound as a yellowish-orange solid (1.18 g, 30%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 7.38 (d, J = 8.68 Hz, 2H, Ph), 7.00 (d, J = 9.12 Hz, 4H, Ph), 6.79 (d, J = 9.16 Hz, 4H, Ph), 6.64 (d, J = 8.68 Hz, 2H, Ph), 3.80 (s, 6H, OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 156.10 (Ph(
3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 156.10 (Ph(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) )–OCH3), 148.53, 140.46, 137.77, 126.77, 122.37, 115.03, 82.03 (Ph()–I), 55.63 (OH3); MS (EI)m/z: 431.
)–OCH3), 148.53, 140.46, 137.77, 126.77, 122.37, 115.03, 82.03 (Ph()–I), 55.63 (OH3); MS (EI)m/z: 431.
4-(2-(4-(Bis(4-methoxyphenyl)amino)phenyl)ethynyl benzal dehyde (7).
4-Ethynylbenzaldehyde (90 mg, 0.69 mmol) was dissolved in THF (10 mL) followed by addition of copper(I)iodide (5 mg, 0.026 mmol), dicholorobis(triphenylphosphine) palladium(II) (8 mg, 0.012 mol) and triethylamine (0.15 mL). Iodo compound 3 (0.27 g, 0.00062 mol) was added from addition funnel as a solution in THF (10 mL) to the above reaction mixture. The reaction mixture was warmed at 30 °C with stirring for 30 min and at 25 °C for 20 h. Analysis of the reaction mixture by TLC indicated the completion of the reaction. After removal of the solvent, the residue was purified on silica gel column (hexane–ethyl acetate: 8/2) to afford the target compound as a yellow solid (0.15 g, 51%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 9. 97 (s, 1H, CO), 7.80 (d, J = 8.24 Hz, 2H, Ph), 7.59 (d, J = 8.24 Hz, 2H, Ph), 7.30 (d, J = 9.16 Hz, 2H, Ph), 7.09–7.05 (m, 4H, Ph), 6.86–6.82 (m, 6H, Ph), 3.79 (s, 6H, OC3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 191.56 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 156.60 (Ph()–CHO), 149.50, 139.94, 135.04, 133.19, 132.79, 131.83, 129.67, 127.39, 118.79, 114.94, 112.59, 94.98 (alkynyl carbon), 87.66 (alkynyl carbon), 55.57 (OH3); MS (EI)m/z: 433.30.
O), 156.60 (Ph()–CHO), 149.50, 139.94, 135.04, 133.19, 132.79, 131.83, 129.67, 127.39, 118.79, 114.94, 112.59, 94.98 (alkynyl carbon), 87.66 (alkynyl carbon), 55.57 (OH3); MS (EI)m/z: 433.30.
(E)-3(4-(4-(Bis(4-methoxyphenyl)amino)phenyl)ethynyl) phenyl)-2-cyanoacetic acid (D1).
Push-pull aldehyde 7 (0.10 g, 0.23 mmol) dissolved in acetonitrile (10 mL) solution was condensed with 2-cyanoacetic acid (19 mg, 0.23 mmol) in the presence of piperidine (29 mg, 0.23 mmol). The above reaction mixture was refluxed for 4 h under nitrogen atmosphere. After cooling to room temperature, the solvent was removed completely. The reaction residue was washed with 2 M aqueous HCl and extracted with CHCl3. The organic phase was collected and dried over Na2SO4. After removal of the solvent, the crude product was purified on the silica gel column (acetic acid–dichloromethane: 1/99) to afford the target compound as a dark red solid (40 mg, 36%).1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.27 (s, 1H, CC–CN), 8.02 (d, J = 8.24 Hz, 2H, Ph), 7.85 (d, J = 8.24 Hz, 2H, Ph), 7.48 (d, J = 8.08 Hz, 2H, Ph), 7.11–7.06 (m, 4H), 6.82–6.88 (m, 6H), 3.79 (s, 6H, OC3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 165.07 (OOH), 155.76 (HC–CN), 150.04 (Ph ()–OCH3), 141.58, 132.01, 131.63, 131.49, 129.77, 129.95, 129.55, 127.70, 127.30, 125.98, 121.95, 118.77, 114.95, 93.31 (alkynyl carbon), 80.64 (alkynyl carbon), 55.59 (OH3); MS (EI)m/z: 500.22; HRMS (ESI, M + 1): m/z cald for C32H25 N2O4 501.1814, found 501.1814.
5-(2-(4-(Bis(4-methoxyphenyl)amino)phenyl)ethynyl)thiophene-2-carbaldehyde (11).
5-(2-(Trimethylsilyl)ethynylthiophene-2-carbaldehyde (0.22 g, 1.61 mole) was dissolved in THF (10 mL) followed by addition of copper(I) iodide (10 mg, 0.053 mol), dicholorobis (triphenylphosphine) palladium(II) (18 mg, 0.026 mol) and triethylamine (0.30 mL). Iodo compound 3 (0.58 g, 1.34 mmol) was added from addition funnel as a solution in THF (10 mL) to the above reaction mixture. The reaction mixture was heated at 30 °C with stirring for 30 min and at 25 °C for 20 h. Monitor of the above reaction mixture by TLC indicated the completion of the reaction. After removal of the solvent, the residue was purified on silica gel column (hexane–ethyl acetate: 8/2) to afford the target compound as yellow solid (0.29 g, 50%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 9. 81 (s, 1H, CO), 7.62 (d, J = 4.12 Hz, 1H, Th), 7.29 (d, J = 8.68 Hz, 2H, Ph), 7.22 (d, J = 4.12 Hz, 1H, Th), 7.09–7.05 (m, 4H), 6.86–6.80 (m, 6H), 3.78 (s, 6H, OC3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 182.53 (O), 156.85 (Ph()–OCH3), 150.00, 143.15, 139.73, 136.31, 134.11, 132.64, 131.67, 127.51, 118.71, 114.80, 111.86, 99.63 (alkynyl carbon), 81.05 (alkynyl carbon), 55.37 (OH3); MS (EI)m/z: 439.16.
(E)-3(4-(4-(Bis(4-methoxyphenyl)amino)phenyl)ethynyl) thiophen-2-yl)-2-cyanoacrylic acid (D2).
Push-pull aldehyde 11 (0.27 g, 0.61 mmol) dissolved in acetonitrile (30 mL) solution was condensed with 2-cyanoacetic acid (0.052 g, 0.61 mmol) in the presence of piperidine (0.052 g, 0.61 mmol). The mixture was refluxed for 4 h under nitrogen atmosphere. The solvent was removed completely after cooling to room temperature; the residue was washed with 2 M aqueous HCl and extracted with CHCl3. The organic phase was collected and dried over Na2SO4. After removal of the solvent, the crude product was purified on the silica gel column (acetic acid/dichloromethane: 1/99) to afford the target compound as a dark red solid (0.124 g, 40%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.25 (s, 1H, C(C)–CN), 7.68 (d, J = 4.12 Hz, 1H, Th), 7.29-7.22 (m, 3H), 7.09 (d, J = 9.16 Hz, 4H, Ph), 6.88–6.79 (m, 6H), 3.79 (s, 6H, OC3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 168.02 (OOH), 156.82 (HC–CN), 149.99 (Ph ()-OCH3), 147.47, 144.51, 143.03, 139.64, 139.00, 135.53, 132.82, 131.76, 127.53, 126.23, 118.35, 115.00, 111.59, 102.12 (alkynyl carbon), 81.29 (alkynyl carbon), 55.62 (OH3); MS (EI)m/z: 506.18; HRMS (ESI, M + 1): m/z cald for C30H23 N2O4S 507.1379, found 507.1376.
5-(5-(2-(4-(Bis(4-methoxyphenyl)amino)ethynyl)thiophene-2-yl)thiophene-2-carbaldehyde (16).
5-(5-Ethynylthiophen-2-yl)thiophene-2-carbaldehyde (0.25 g, 1.14 mmol) was dissolved in THF (10 mL), followed by addition of copper(I) iodide (10 mg, 0.05 mmol), dicholorobis(triphenylphosphine)palladium(II) (70 mg, 0.09 mmol) and triethylamine (0.50 mL). Iodo compound 3 (0.50 g, 1.16 mmol) was added from addition funnel as a solution in THF (10 mL). The reaction was heated with stirring at 30 °C for 30 min and then at 25 °C for 20 h. Analysis of the reaction by TLC indicated the completion of the reaction. After removal of the solvent, the residue was purified on silica gel column chromatography to afford the target compound 16 (0.30 g, 51%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 9. 84 (s, 1H, CO), 7.63 (d, J = 4.12 Hz, 1H, Th), 7.26 (d, J = 8.75 Hz, 2H, Ph), 7.20–7.22 (m, 2H), 7.11 (d, J = 4.12 Hz, 1H, Th), 7.05 (d, J = 9.60 Hz, 4H, Ph), 6.80–6.86 (m, 6H), 3.78 (s, 6H, OC3); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ = 182.53 (HO), 156.64 (Ph ()–OCH3), 149.39, 146.50, 141.85, 139.87, 137.43, 136.25, 132.44, 132.22, 127.35, 126.16, 125.87, 124.40, 118.85, 114.94, 112.61, 96.70 (triple bond), 80.81 (triple bond), 55.13 (OH3); MS (EI)m/z: 521.
(E)-3-(5-(5-(2-(4-(Bis(4-methoxyphenyl)amino)phenyl)ethynyl)thiophene-2-yl)thiophene-2-yl)-2-cyanoacrylic acid (D3).
Aldehyde
16 (0.20 g, 0.38 mmol) dissolved in acetonitrile (10 mL) solution was condensed with 2-cyanoacetic acid (032 mg, 0.38 mmol) in the presence of piperidine (36 mg, 0.42 mmol). The mixture was refluxed for 4 h under N2. After cooling to room temperature, the solvent was removed completely. The residue was washed with 2 M aqueous HCl and extracted with CHCl3. The organic phase was collected and dried over Na2SO4. After removal of the solvent, the crude product was purified on the silica gel column (1% acetic acid in DCM) to afford the target compound D3 as a dark red solid (80 g, 36%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.25 (s, 1H, CC–CN), 7.70–7.68 (m, 1H), 7.53–7.50 (m, 1H), 7.14–6.99 (m, 6H), 6.75–6.88 (m, 8H), 3.77 (s, 6H, OC3); 13C NMR (100 MHz, THF-d8, 25 °C, TMS): δ = 167.89 (OOH), 156.50 (HC–CN), 149.17 (Ph()–OCH3), 146.74, 140.19, 139.98, 136.02, 134.60, 132.50, 132.46, 130.98, 128.88, 127.28, 127.12, 119.07, 118.87, 116.80, 114.88, 112.75, 96.96 (triple bond), 81.18 (triple bond), 55.49 (OH3); MS (EI)m/z: 588; HRMS (ESI, M+1): m/z cald for C34H25 N2O4S2 589.1256, found 589.1241.MS (EI) m/z: 684; HRMS (ESI, M + 1): m/z cald for C34H25 N2O4Se2 685.0145, found 685.0157
Solar cell fabrication
The transparent conducting glass (SnO2:F, FTO, 15 Ω per square, Solaronix) electrodes into a 0.04 M TiCl4 aqueous solution for 1 h at 70 °C until the TiCl4 was hydrolyzed, then washing the electrodes with distilled water to remove residual TiCl4, and TiO2 (Ti–Nanoxide T/SP, Solaronix) paste were deposited on the treated transparent conducting glass with screen printing method and sintered at 500 °C in air for 30 min and then cooled to room temperature. 400 nm TiO2 scratter layer (DSL 18NR-AO) were deposited on the TiO2 film and sintered at 500 °C in air for another 30 min. Then the TiO2 (7 + 5 μm) film electrode were soaked in an chloroform solution containing 0.5 mM organic dye for 12 h at room temperature and then washed with ethanol and dried in dry air. A sandwich cell was prepared using the dye-sensitized electrode as the working electrode and a platinum-coated conducting glass electrode as the counter electrode. The counter electrode used was a 400 Å Pt fabricated by e-beam evaporation on a commercial indium tin oxide glass. The two electrodes separated by 60 μm thermal-plastic Suryln spacers were bonding together, and the electrolyte (EL-HPE) was introduced between the electrodes by capillary action.
Acknowledgements
Nanyang Technological University (RG54/07 and SUG41/06) and the Ministry of Education, Singapore (ARC24/07, no. T206B1218RS) are gratefully acknowledged.
References
-
(a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS;
(b) K. Hara, Z. S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476–15482 CrossRef CAS;
(c) E. M Barea, R. Caballero, F. Fabregat-Santiago, P. De La Cruz, F. Langa and J. Bisquert, ChemPhysChem, 2009, 11, 245–250;
(d) L. M. Peter and K. G. U. Wijayantha, Electrochim. Acta, 2000, 45, 4543–4551 CrossRef CAS;
(e) K. Hara, H. Sugihara, Y. Tachibana, A. Islam, M. Yanagida, K. Sayama, H. Arakawa, G. Fujihashi, T. Horiguchi and T. Kinoshita, Langmuir, 2001, 17, 5992–5999 CrossRef CAS;
(f) P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS;
(g) A. J. Frank, N. Kopidakis and J. van de Lagemaat, Coord. Chem. Rev., 2004, 248, 1165–1179 CrossRef CAS;
(h) Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Y. Han, Jpn. J. Appl. Phys., 2006, 45, L638–640 CrossRef CAS;
(i) M. K. Nazeeruddin and M. Grätzel, Struct. Bonding, 2007, 123, 113–175 CAS.
-
(a) M. Alebbi, C. A. Bignozzi, T. A. Heimer, G. M. Hasselmann and G. J. Meyer, J. Phys. Chem. B, 1998, 102, 7577–7581 CrossRef CAS;
(b) G. Sauve, M. E. Cass, S. J. Doig, I. Lauermann, K. Pomykal and N. S. Lewis, J. Phys. Chem. B, 2000, 104, 3488–3491 CrossRef CAS;
(c) M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphrybaker, E. Muller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
-
(a) F. C. Krebs and M. Biancardo, Sol. Energy Mater. Sol. Cells, 2006, 90, 142–165 CrossRef CAS;
(b) K. Ikegami, Curr. Appl. Phys., 2006, 6, 813–816 CrossRef;
(c) Y. H. Jiang, Y. C. Wang, J. L. Hua, J. Tang, B. Li, S. X. Qian and H. Tian, Chem. Commun., 2010, 46, 4689–4691 RSC.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS and references therein.
- M. K. Mishra, R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS and references therein.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Han, Jpn. J. Appl. Phys., 2006, 45, L638–640 CrossRef CAS.
- Y. T. Chang, S. L. Hsu, G. Y. Chen, M. H. Su, T. A. Singh, E. W. G. Diau and K. H. Wei, Adv. Funct. Mater., 2008, 18, 2356–2365 CrossRef CAS.
- S. Z. Zhang, L. D. Sun, H. Tian, H. Y. Liu, J. F. Wang and C. H. Yan, Chem. Commun., 2009, 1766–1768 RSC.
- K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597–606 CrossRef CAS.
- Z. Wang, Y. Cui, K. Hara, Y. Dan-oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS.
- Z. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224–7230 CrossRef CAS.
- K. Sayama, K. Hara, N. Mori, M. Satsuki, S. Suga, S. Tsukagoshi, Y. Abe, H. Sugihara and H. Arakawa, Chem. Commun., 2000, 1173–1174 RSC.
- K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, J. Phys. Chem. B, 2002, 106, 1363–1371 CrossRef CAS.
- X. Ma, J. Hua, Y. Jin, F. Meng, W. Zhan and H. Tian, Tetrahedron, 2008, 64, 345–350 CrossRef CAS.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218–12219 CrossRef CAS.
- L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813–818 CrossRef.
- K. Hara, T. Sato, R. Katoh, A. Furube, T. Yoshihara, M. Murai, M. Kurashige, S. Ito, A. Shinpo, S. Suga and H. Arakawa, Adv. Funct. Mater., 2005, 15, 246–252 CrossRef CAS.
- Z.-S. Wang, F.-Y. Li and C.-H. Huang, Chem. Commun., 2000, 2063–2064 RSC.
- Q. H. Yao, F. S. Meng, F. Y. Li, H. Tian and C. H. Huang, J. Mater. Chem., 2003, 13, 1048–1051 RSC.
- M. Velusamy, K. R. J. Thomas, J. T. Lin, Y. C. Hsu and K. C. Ho, Org. Lett., 2005, 7, 1899–1902 CrossRef CAS.
- T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. Anderson, X. Ai, T. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806–1812 CrossRef CAS.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245–2247 RSC.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J. H. Yum, S. Fantacci, F. D. Angelis, D. D. Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS.
- D. Kim, J. K. Lee, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 1913–1922 CrossRef CAS.
- H. Choi, J. K. Lee, K. Songs, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 3115–3121 CrossRef CAS.
- R. Chen, X. Yang, H. Tian and L. Sun, J. Photochem. Photobiol., A, 2007, 189, 295–300 CrossRef CAS.
- G. Zhang, H. Bala, Y. Cheng, D. Shi, X. Lv, Q. Yu and P. Wang, Chem. Commun., 2009, 2198–2200 RSC.
-
(a) J. S. Song, F. Zhang, C. H. Li, W. L. Liu, B. S. Li, Y. Huang and Z. S. Bo, J. Phys. Chem. C, 2009, 113, 13391–13397 CrossRef CAS;
(b) M. Liang, W. Xu, F. Cai, P. Chen, B. Peng, J. Chen and Z. Li, J. Phys. Chem. C, 2007, 111, 4465–4472 CrossRef CAS;
(c) Y. Shibano, T. Umeyama, Y. Matano and H. Imahori, Org. Lett., 2007, 9, 1971–1974 CrossRef CAS;
(d) N. S. Baek, J. H. Yum, X. X. Zhang, H. K. Kim, M. K. Nazeeruddin and M. Gratzel, Energy Environ. Sci., 2009, 2, 1082–1084 RSC.
-
(a) N. Satoh, T. Nakashima and K. Yamamoto, J. Am. Chem. Soc., 2005, 127, 13030–13038 CrossRef CAS;
(b) N. Satoh, J. S. Cho, M. Higuchi and K. Yamamoto, J. Am. Chem. Soc., 2003, 125, 8104–8105 CrossRef CAS.
- Q. Wang, S. M. Zakeeruddin, J. Cremer, P. Bauerle, R. Humphry-Baker and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 5706–5713 CrossRef CAS.
- Y. J. Chang and T. J. Chow, Tetrahedron, 2009, 65, 9626–9632 CrossRef CAS.
- J. H. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nüesch, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320–10321 CrossRef CAS.
- X. M. Ma, W. J. Wu, Q. Zhang, F. L. Guo, F. S. Meng and J. L. Hua, Dyes Pigm., 2009, 82, 353–359 CrossRef CAS.
- D. Geppert and R. de Vivie-Riedle, Springer Ser. Chem. Phys., 2007, 88, 258 Search PubMed.
- D. P. Hagberg, J. H. Yum, H. J. Lee, F. D. Angelis, T. Marinado, K. M. Karlsson, R. Humphry-Baker, L. C. Sun, A. Hagfeldt, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2008, 130, 6259–6266 CrossRef CAS.
- A. A. Bothner-By, J. Dadok, T. E. Johnson and J. S. Lindsey, J. Phys. Chem., 1996, 100, 17551–17557 CrossRef CAS.
- J. L. Fillaut, J. Perruchon, P. Blanchard, J. Roncali, S. Golhen, M. Allain, M.-Z. Anna, I. V. Kityk and B. Sahraoui, Organometallics, 2005, 24, 687–695 CrossRef CAS.
- A. F. Michael, P. L. Olivia and I. S. David, Org. Lett., 2008, 10, 4979–4982 CrossRef CAS.
- J. T. Lin, P. C. Chen, Y. S. Yen, Y. C. Hsu, H. H. Chou and M. C. P. Yeh, Org. Lett., 2009, 11, 97–100 CrossRef CAS.
- R. Li, X. Lv, D. Shi, D. Zhou, Y. Cheng, G. Zhang and P. Wang, J. Phys. Chem. C, 2009, 113, 7469–7710 CrossRef CAS.
- J. O. Morley and D. Push, J. Chem. Soc., Faraday Trans., 1991, 87, 3009–3013 RSC.
- Lee, V. Suryanarayanan, J. H. Huang, K. R. J. Thomas, J. T. Lin and K. C. Ho, Electrochim. Acta, 2009, 54, 4123–4130 CrossRef CAS.
-
(a) A. Ehret, L. Stuhl and M. T. Spitler, J. Phys. Chem. B, 2001, 105, 9960–9965 CrossRef CAS;
(b) K. Han, X. Z. Lu, J. H. Xu, G. P. Zhou, S. H. Ma, W. C. Wang, Z. G. Cai and J. Y. Zhou, J. Phys. D: Appl. Phys., 1997, 30, 2923–2927 CrossRef CAS;
(c) S. Wenger, P. A. Bouit, Q. L. Chen, J. Teuscher, D. D. Censo, R. Humphry-Baker, J. E. Moser, J. L. Delgado, N. Martin, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2010, 132, 5164–5169 CrossRef CAS;
(d) J. B. Asbury, N. A. Anderson, E. Hao, X. Ai and T. Lian, J. Phys. Chem. B, 2003, 107, 7376–7386 CrossRef CAS;
(e) J. B. Asbury, E. Hao, Y. Wang, H. N. Ghosh and T. Lian, J. Phys. Chem. B, 2001, 105, 4545–4557 CrossRef CAS;
(f) J. B. Asbury, E. Hao, Y. Wang and T. Lian, J. Phys. Chem. B, 2000, 104, 11957–11964 CrossRef CAS.
-
(a) Q. Wang, J. E. Moser and M. Gra1tzel, J. Phys. Chem. B, 2005, 109, 14945–14953 CrossRef CAS;
(b) A. Burke, S. Ito, H. Snaith, U. Bach, K. Kwiatkowski and M. Gratzel, Nano Lett., 2008, 8, 977–981 CrossRef CAS;
(c) Z. J. Ning, Q. Zhang, H. C. Pei, J. F. Luan, C. G. Lu, Y. P. Cui and H. Tian, J. Phys. Chem. C, 2009, 113, 10307–10313 CrossRef CAS;
(d) K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597–606 CrossRef CAS;
(e) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS.
-
(a) A. J. Frank, N. Kopidakis and J. van de Lagemaat, Coord. Chem. Rev., 2004, 248, 1165 CrossRef CAS;
(b) B. A. Gregg, J. Phys. Chem. B, 2001, 105, 1422 CrossRef CAS;
(c) M. Gratzel, J. Photochem. Photobiol., C, 2003, 4, 145 CrossRef CAS;
(d) M. K. Wang, C. Gratzel, S. J. Moon, R. Humphry-Baker, N. Rossier-Iten, S. M. Zakeeruddin and M. Gratzel, Adv. Funct. Mater., 2009, 19, 2163 CrossRef CAS.
- S. Hwang, J. H. Lee, C. Park, H. Lee, C. Kim, M. H. Lee, W. Lee, J. Park, K. Kim and N. G. Park, Chem. Commun., 2007, 4887–4889 RSC.
-
(a) J. H. Yum, S. J. Moon, R. Humphry-Baker, P. Walter, T. Geiger, F. Nuesch, M. Gräetzel and M. D. K. Nazeeruddin, Nanotechnology, 2008, 19, 424005-1–424005-6;
(b) J. H. Yum, S. R. Jang, R. Humphry-Baker, M. Grätzel, J. Cid, T. Torres and M. K. Nazeeruddin, Langmuir, 2008, 24, 5636–5640 CrossRef CAS.
-
(a) R. Ditchfield, W. J. Herhe and A. J. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS;
(b) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS;
(c) M. Cossi, N. Rega, G. Scalmani and V. J. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- Y. X. Weng, L. Li, Y. Liu, L. Wang and G. Z. Yang, J. Phys. Chem. B, 2003, 107, 4356–4363 CrossRef CAS.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed;
(b)
R. Dennington, T. Keith, J. Millam, K. Eppinnett, W. L. Hovell and R. Gilliland, GaussView, Semichem, Shawnee Mission, KS, 2003 Search PubMed.
-
(a)
S. I. Gorelsky, AOMix Program for Molecular Orbital Analysis, York University, Toronto, 1997 Search PubMed;
(b) S. I. Gorelsky and B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, electrochemistry spectra and additional figures. See DOI: 10.1039/c0nj00653j |
| ‡ J. L. Song and P. Amaladass contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.