Hierarchically structured carbon replica of hybrid layered double hydroxide†
Vanessa
Prévot
*ab,
Erwan
Géraud
ab,
Thomas
Stimpfling
ab,
Jaafar
Ghanbaja
c and
Fabrice
Leroux
ab
aCNRS, UMR 6002, Laboratoire des Matériaux Inorganiques, F-63177, Aubière, France. E-mail: vanessa.prevot@univ-bpclermont.fr; Fax: +33 4 73 40 71 08; Tel: +33 4 73407104
bClermont Université, Université Blaise Pascal, BP 10448, F-63000, Clermont-Ferrand, France
cUniversité Henri Poincaré, Faculté des Sciences et Techniques, Service Commun de Microscopies Electroniques et Microanalyses X, Bd des Aiguillettes, BP 239, 54506 Vandoeuvre-Lès-Nancy
First published on 11th October 2010
Abstract
A hierarchically structured carbon replica with a tri-modal distribution of porosity was prepared by pyrolysis of a three-dimensionally ordered macroporous (3-DOM) organic–inorganic hybrid layered double hydroxide (LDH). By an original inverse opal method involving LDH platelet regeneration, macroporous Mg2Al–vinyl benzene sulfonate (VBS) was synthesized and subsequently submitted to a moderate thermal treatment to induce the VBS in situpolymerization. Successive steps of carbonization and demineralization were conducted to yield to a carbon replica. A thermal treatment under inert atmosphere ensures the carbonization of the organic part and the simultaneous decomposition of the LDH into oxide and mixed oxide particles. The latter acts as a porogen agent during the acid-leaching step, which induces the removal of the inorganic part. The samples upon carbonization and the capacitive properties of the resulting carbon replica were thoroughly characterized using powder X-ray diffraction (PXRD), Fourier-transform infra-red (FTIR) and 13C nuclear magnetic resonance (NMR) spectroscopy, field-emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and Brunauer–Emmet–Teller (BET) analysis of the nitrogen adsorption isotherm. 3-DOM carbon replicas exhibit poor capacitive behaviour in aqueous electrolyte, explained by a high ionic resistivity present in the porosity. Even though this electrical disruptive effect can be explained on the basis of the macroporosity of the disconnected carbon 3-DOM, this allows us to envisage other applications for this approach in order to obtain tri-modal carbon-based materials.
Introduction
Carbon’s unique physical and chemical properties make it an excellent candidate for many applications. Carbon is extensively used as an electrode material (lithium-ion batteries, fuel cells, supercapacitors), adsorbents and as a catalyst support. Because nanostructured materials often display a performance enhancement, many advanced synthetic methods have been developed to produce porous carbon materials with suitable properties.1–3In the literature, the template carbonization method is one of the most powerful and promising ways of producing well-defined nanostructured carbon materials. This method consists of the calcination under an inert atmosphere of a carbon source in the presence of a template. Subsequent template removal then liberates the carbonaceous materials. Two major classes of templates can be distinguished:3–5 the soft and hard templates. The former relies on the supramolecular assembly of block copolymers4,6 (such as polystyrene-b-poly(4-vinylpyridine), PS-P4VP, and poly(ethylene oxide)-b-poly(propylene oxide)-b-(polyethylene oxide), PEO–PPO–PEO), which further interact through hydrogen bonding with carbon precursors, typically resorcinol–formaldehyde, phloroglucinol–formaldehyde, resol and carbohydrates.
In the hard template approach, organic or inorganic nanostructures possessing a well-defined porosity are used for replication synthesis. Microporous compounds such as zeolites are efficient templates7–11 to synthesize microporous carbons which can display ultrahigh surface area (3600 m2 g−1).10 When using ordered mesoporous silica with different structures (MCM-41, Al-HMS, MCM-48, SBA-15, SBA-1, FDU-5, etc.) as the hard porous template,5 ordered mesoporous carbon with a variety of structures and symmetries were prepared. In this case, the macroscopic morphology and the ordered structure of the template were maintained. Layered materials such as clays or anionic synthetic clays so called layered double hydroxides (LDHs) have also been investigated to prepare porous graphite-like carbons or carbon–mineral nanocomposites from intercalated carbon precursors12–15 such as sucrose, polymer or monomer, including poly(furfuryl alcohol), acrylonitrile, vinyl acetate and vinyl benzene sulfonate. Schwarz et al.16 emphasized that the chemical composition and the water content of the inorganic template (taeniolite) are the dominant factors for the development of the micropore within the carbon. The templates involved in the present work are LDH, also called hydrotalcite-like compounds.17 They possess a layered structure with the ideal formula: [MII1−xMIIIx(OH)2]x+[Xq−x/q(H2O)n]x− where MII and MIII are metal cations. The substitution of some of the divalent cations by trivalent cations in the brucite-like layers creates a positive charge which is compensated by the presence of anions Xq− in the interlayer domain.
One of the main advantages of the LDH structure is that their chemical composition can be easily tuned. In particular, the possible intercalation of organic anions can lead to hybrid materials, therefore extending the field of application of LDH as supported catalysts,18 multifunctional materials19 and compatible inorganic fillers.20 Carbonization under inert conditions of such hybrid layered compounds, followed by the removal of the inorganic part by acid demineralization, can give access to a variety of interesting carbonaceous materials. Both the nature of the LDH layers and the confined organic parts can affect the textural and electrochemical properties of the carbonaceous products. Leroux et al.14 showed that the intercalation of a monomer and its subsequent in situpolymerisation, carbonisation and demineralisation give rise to carbons with higher microporous and mesoporous contributions than that obtained by equivalent treatment using simple polymer sequestration only.
In order to improve the diffusion and the ions transport inside carbons, as well as to achieve adsorption of large molecules in an open structure, there is great interest in combining the micro and/or mesoporosity with macroporosity.21 Specifically, whereas macropores can facilitate transport, the smaller pores provide a large surface area on which reactions can take place.
A common template method to access three-dimensionally ordered macroporous (3-DOM) solids involves the use of a sacrificial colloidal crystal (opal) which is a closely packed array of mono-dispersed silica or polymer beads.22 3-DOM carbons with a hierarchical porosity were prepared by infiltration of a carbon source into the interstitial voids of the array, the macroporosity being subsequently generated by removal of the beads.23 For instance, well-ordered macroporous carbons with a microporous framework (S = 408 m2g1) can be obtained by carbonizing an aqueous solution of sucrose infiltrated into a SiO2 colloidal crystal.24 Recently, Kanamura and co-worker reported25,26 on the preparation of 3-DOM carbons with walls composed of mesosized hollow spheres using a colloidal crystal template containing both polystyrene (PS) beads (450 nm) and colloidal silica (<50 nm). Another strategy is to use a pre-fabricated inverse silica opal as the template. Carbon is then deposited using chemical vapour deposition, before dissolution of the silica template in acidic solution.27
In our previous work, we described how inverse opals or 3-DOM LDH28,29 can be prepared using a PS colloidal crystal templating method. We assumed that the LDH macroporous architecture might be maintained during the successive steps of carbon production, giving rise to carbon with a hierarchically structured porosity. Herein, we report the template synthesis of nanostructured carbon through carbonization of an organic precursor confined in the bidimensional interlayered space of the 3-DOM LDH materials. Both the endo- and the exo-templating approaches30 are used successively, the PS crystal acting as an endotemplate, whereas the LDH can be considered as an exotemplate allowing the incorporation of the carbon source into the interlamellar domain. The procedure as depicted in Fig. 1 involves three main steps. The first is the preparation of 3-DOM vinyl benzene sulfonate (VBS) intercalated LDH by calcination–regeneration. In the second step, the in situpolymerization of VBS in the 3-DOM LDH is carried out under moderate thermal treatment. In the third step, carbon is formed by carbonization and the template residues are removed by exposure to an acidic solution.
 | ||
| Fig. 1 Illustration of the synthetic method used for the preparation of a carbon replica. | ||
Since the large specific surface area is in this case achieved in association with a pore size distributed at different scales (which should be an advantage for ion transport), we studied the capacitive behavior of the 3-DOM carbon replica by cyclic voltammetry in Na2SO3 aqueous electrolyte. Indeed, in the course of electrochemical capacitive energy storage based on supercapacitor systems,31 numerous carbon materials, in particular those with a bi-modal pore size distribution,32 have been tested for such applications as electric double layer capacitors (EDLCs). A carbon replica reference (denoted C-carbon), obtained from Mg2Al-VBS but without the 3-DOM arrangement, was studied for comparison.
Experimental
All the chemicals used were of reagent grade and were used as supplied, except for the styrene monomer used for the synthesis of colloidal PS spheres, which was first distilled to remove any inhibitors.Synthesis of 3-DOM VBS intercalated Mg2Al–LDH hybrid
The first step of the synthetic process (Fig. 1) was the preparation of macroporous Mg2Al–(VBS monomer) nanohybrids (denoted 3-DOM Mg2Al–VBS), that will subsequently be used as starting materials to produce carbonaceous compounds. Suspension of monodisperse PS beads (∅ = 710 ± 10 nm) was prepared using “emulsifier-free” emulsion polymerization and packed into 3DO colloid crystals by centrifugation. The Mg2Al–LDH coprecipitation around the PS beads was conducted by successive impregnations as previously reported.28,29 The Mg2Al–PS composite thus obtained was calcined in a tubular furnace in an air flow at 673 K for 12 h. After calcination, the resulting 3-DOM Mg,Al mixed oxides (hereafter noted as 3-DOM MO) were regenerated by immersion into 50 mL of an aqueous solution containing VBS sodium salt (0.1 M) for 1 day under a nitrogen atmosphere in order to minimize the contamination with atmospheric CO2. The sample thus obtained was filtered, washed with deionised water and allowed to dry at room temperature in air overnight. Then, the in situpolymerization of intercalated VBS molecules was thermally induced in air at different temperatures ranging from 180° to 220 °C. The Mg2Al–(VBS polymer) hybrids obtained are hereafter denoted 3-DOM Mg2Al–VBSp.Preparation of carbonaceous materials
The 3-DOM Mg2Al–VBSp hybrid material was converted into 3-DOM carbon by calcination in a tubular furnace under nitrogen flow (50 mL min−1). The temperature was increased at a rate of 5 K min−1 to 873 K for 8 h and cooled down to room temperature. The dissolution of the inorganic part was carried out by acid leaching using hydrochloric acid (1 M, 72 h). The resulting black powders were filtered and then washed with deionized water twice.For comparison, carbon was also prepared from standard coprecipitated Mg2Al phase at pH 10.0 ± 0.1 using an automated titrator. The addition of the metallic salt solution was completed in 4 h, and the mixture was then left to age for 48 h, washed three times with deionized water and allowed to dry at room temperature overnight. The subsequent procedures, i.e. VBS intercalation, VBS in situpolymerization, carbonization and leaching to produce carbon material (hereinafter denoted C-carbon), were similar to those described above.
Characterization of materials
The PS beads sizes were measured with a Malvern Zetasizer (Nano ZS) from dilute suspensions. Chemical analyses were performed by inductively coupled plasma atom emission spectroscopy at the Vernaison Analysis Center of CNRS (France). Powder X-ray diffraction (PXRD) experiments patterns were carried out with a Siemens D501 X-ray diffractometer using CuKα radiation (λ = 1.5415 Å) and fitted with a graphite back-end monochromator. Diagrams were acquired from 2° to 70° in 2θ using a step of 0.08° and a counting time per step of 4 s. In situX-ray diffraction patterns were obtained by an X'Pert Pro Philips diffractometer with a diffracted-beam graphite monochromator using a Cu Kα radiation source equipped with a high-temperature chamber (Anton Paar HTK-16). Diffractograms were recorded in the 2θ range of 5–70° with a step of 0.013° and a counting time per step of 20 s.FTIR spectra were collected on a Perkin Elmer 16PC spectrophotometer using KBr pellets. SEM characteristics of the samples were imaged by a Zeiss supra 55 FEG-VP operating at 3 keV. SEM samples were deposited onto a conductive carbon paper adhesive and were covered with a thin layer of gold to make them conductive.
Transmission electron microscopy (TEM) images were taken using a Philips CM20 microscope at an acceleration voltage of 200 kV. Samples were dispersed in ethanol and then one droplet of the suspension was applied to a 400 mesh holey carbon-coated copper grid and left to dry in air.
Nitrogen adsorption measurements at liquid nitrogen temperature were performed using a FISON SP1920 sorption analyzer. The specific surface area was calculated by applying the Brunauer–Emmett–Teller (BET) equation, and the pore size distribution was calculated by the Barrett–Joyner–Halenda (BJH) method using the desorption branch. Prior to the experiments, samples were degassed under a vacuum at 393 K for 12 h.
13C (I = 1/2) solid-state NMR experiments were performed with a 300 Bruker spectrometer at 75.47 MHz. The experiments were carried out using magic-angle spinning (MAS) at 10 kHz and a 4 mm diameter size zirconia rotor. 13C spectra obtained by proton-enhanced cross-polarization (CP) are referenced to the carbonyl of the glycine calibrated at 176.03 ppm.
Thermogravimetric analyses (TGA) were recorded on a Setaram TG-DTA 92 thermogravimetric analyser in the temperature range 289–1273 K, with a heating rate of 5 K min−1.
Electrochemical measurements
The electrodes were obtained by mixing active material (80%) acetylene black (10%) and poly(vinylidene fluoride) (10%) in ethanol. Both electrodes of comparable mass (in the range of 0.3–2.5 mg) were deposited on stainless steel 316 L disk (∅ = 12 mm) and electrically isolated from each other by a glassy fibrous separator used as an electrolyte reservoir. The materials were tested in symmetric two-electrode capacitors using a stainless steel Swagelok®cell, the inner tube being isolated by a polypropylene membrane. The aqueous electrolyte was prepared with Na2SO3 (1.0 mol L−1, pH 9.7). The electrochemical characterizations were performed by cyclic voltammetry with a VSP multichannel potentiostat/galvanostat (BioLogic, EC-Lab V9.4 Software). The voltammograms were recorded in the range from −0.5 to +0.6 V. The scan rates were 0.1, 2, 5, 10, 50, and 100 mV s−1. The capacitance was calculated from the recorded current per unit of active mass (I, in units of A g−1) in the investigated potential domain divided by the sweep rate value (dV/dt, in units of V s−1), following the formula C = 2Ic/(dV/dt) in units of A s V−1 g−1 (≡ F g−1), where Ic is the intensity measured during the cathodic sweep, and dV/dt is the potential step. For the galvanostatic mode (constant current), the electrode materials were cycled within the optimized potential domain ΔV. In this case, C = 2I(dt/dV)exp where I is a constant current (expressed in units of A g−1) and (dt/dV)exp is the experimental slope of the t–V curve. The I–V profile for acetylene black deposited on a stainless steel electrode was determined for the tested electrolytes with small capacitances (<2 F g−1). Complex impedance spectroscopy was performed on the electrochemical cell at a fixed potential imposed 5 min before applying a peak-to-peak amplitude of 20 mV and a frequency domain from 5 mHz to 200 kHz to access the equivalent serial resistance (ESR).Results and discussion
3-DOM Mg2Al–VBS hybrid phase
The 3-DOM Mg2Al–VBS hybrid LDH was prepared using the calcination reconstruction process involving a PS opal/carbonate intercalated Mg2Al phase composite.33 This latter was synthesized as previously reported through an inverse opal approach, in which a colloidal crystal of close-packed monodispersed PS spheres (∅ = 710 nm) was used as an exo-template.28,29Fig. 2 clearly shows both the well-ordered colloidal structure with close-packed PS spheres and the macroporous structure of the 3-DOM MO obtained after the two successive impregnation steps and removal of the polymeric template by calcination. The template elimination induces a shrinkage of the porous architecture; the average diameter of the voids being 30–35% smaller than that of the initial PS sphere used (Table 1). After the LDH regeneration step in VBS aqueous solution, it should be emphasized that the ordered arrangement of the macroporous structure is more limited due to some defects which may be induced by structural collapses of the inverse opal during the reconstruction process. Indeed, even though the LDH reconstruction process is debatable, several recent publications are in favor of a mechanism based on local dissolution–precipitation.34 This phenomenon in the presence of VBS induces a partial collapse of the spherical pores, which are not as highly regular as in the 3-DOM MO. The XRD patterns of the samples at the different steps of the process are displayed in Fig. 3. | ||
| Fig. 2 SEM micrographs of (a) PS starting crystal, (b) 3DOM MO obtained after calcination of (a) and (c) regenerated 3 DOM Mg2Al–VBS by rehydration of (b) in VBS solution. | ||
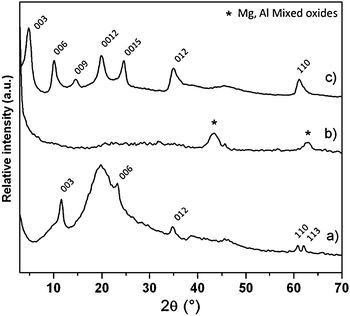 | ||
| Fig. 3 PXRD pattern of (a) the Mg2Al/PS composite, (b) the 3DOM-MO obtained by calcination of (a) at 673 K and (c) the regenerated Mg2Al–VBS monomer by rehydration of (b) in VBS solution. | ||
| Matrix | Chemical formulaa | d-spacing (nm) | BET surface area (m2 g−1) | Macropore diameter (nm)b | Wall thickness (nm)b |
|---|---|---|---|---|---|
| a Calculated from chemical analysis. b Estimated from the SEM image. | |||||
| 3-DOM MO | 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MgO + 0.5MgAl2O4 MgO + 0.5MgAl2O4 |
— | 95 | 480 ± 15 | 65 ± 5 |
| 3-DOM Mg2Al–VBS |
Mg1.9Al(OH)5.8(C8H7SO3)1.1·2.3H2O |
1.84 | 8 | 450 ± 20 | 70 ± 15 |
| 3-DOM Mg2Al–VBSp | Mg1.9Al(OH)5.8(C8H7SO3)1.1 | 1.61 | — | 390 ± 10 | 55 ± 20 |
| 3-DOM carbon | — | — | 1205 | 365 ± 10 | 40 ± 15 |
After impregnation into the colloidal crystal, the characteristic diffraction lines of a hydrotalicte-like (00l and hkl) compound are present, though partially masked by a large hump resulting from the PS array.
The calcination gives rise to LDH decomposition, which is illustrated by the presence of typical reflections of Mg/Al spinel-like phase and MgO (periclase). After rehydration, the regenerated matrix displays the typical diffraction peaks of the LDH structure indexed in a hexagonal lattice with a R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m rhombohedral symmetry.
m rhombohedral symmetry.
The effective intercalation of the VBS monomer into the LDH interlamellar space is confirmed by the position of the (00l) lines corresponding to a basal spacing of 1.84 nm.
This distance is comparable to the values observed for the VBS intercalated Zn2Al- and Ca2Al-matrices of 1.82 and 1.77 nm respectively.35,36 This is in agreement with the arrangement of the VBS molecules in a bilayer. One should note the absence of carbonate anion contamination, thus underlining the efficiency of the procedure. However, the broadness of the (00l) and (110) lines suggests the presence of disorder in the stacking sequence (turbostratic effect) and within the layer structure, respectively. Note that despite the VBS intercalation into the layered structure, the 3-DOM architecture is still maintained and the wall thickness of the macroporous structure remains almost constant at 70 ± 15 nm (Fig. 2).
The chemical analysis confirms that the composition of the hybrid LDH phase obtained is close to the expected one (Table 1), thus underlining the efficiency of the procedure in spite of its successive steps. The presence of the VBS molecules in hybrid materials is also confirmed by FTIR spectroscopy (Fig. 4).
 | ||
| Fig. 4 FTIR spectra of (a) VBS molecule, (b) the Mg2Al–VBS and (c) the Mg2Al–VBSp obtained by thermal treatment at 493 K of (b). | ||
For the 3-DOM Mg2Al–VBS, the characteristic absorption bands of the VBS monomer molecules ν(S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)as, ν(SO)s and S–Ph appear respectively at 1171, 1036 and 1034 cm−1. Compared to the single VBS molecules, the downshift in frequency observed indicates a weakening of the strength of the SO bond due to electrostatic interaction between the anionic group of the organic molecules and the positively charged inorganic layers.
O)as, ν(SO)s and S–Ph appear respectively at 1171, 1036 and 1034 cm−1. Compared to the single VBS molecules, the downshift in frequency observed indicates a weakening of the strength of the SO bond due to electrostatic interaction between the anionic group of the organic molecules and the positively charged inorganic layers.
In situ polymerization
In situ polymerization of VBS monomers was induced by heating the 3-DOM Mg2Al–VBS sample in air. The process was followed by both XRD and 13C NMR in CP-MAS conditions (Fig. 5).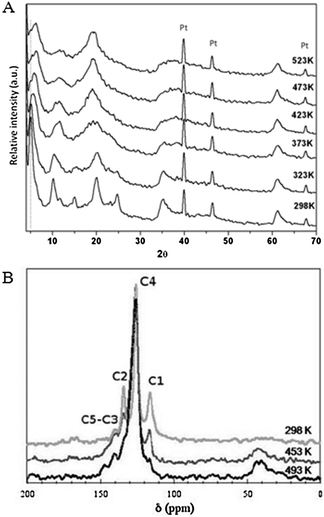 | ||
| Fig. 5 (A) In situXRD pattern of the hybrid phase Mg2Al–VBS recorded every 50 K from room temperature up to 523 K and (B) 13C CP-MAS NMR spectra of the hybrid phase Mg2Al–VBS recorded before and after thermal treatment at 453 K and 493 K. | ||
The XRD pattern displays a shift of the (00l) diffraction lines upon thermal treatment, which indicates a contraction of the interlamellar space from 1.84 nm to 1.61 nm. This was attributed to dehydration and conformational change in the VBS molecules arrangement, caused by opening of the C–C bond and polymerization confined into the interlayer space.35
To get further insight into the monomer polymerization mechanism, 13C NMR spectra were recorded in CP-MAS conditions at different temperatures (Fig. 5b). At room temperature, the different peaks observed for the 3-DOM Mg2Al–VBS sample can be attributed to each carbon of the molecule as follows: C1H2–C2H–C3(C44H4)C5–SO3−.35 However, some of the resonance peaks are shifted compared with the VBS molecule due to a shielding effect for C2, C3 and C5carbon atoms and a de-shielding effect for C1. Such modifications can be explained by the electrostatic interactions between the VBS anionic group and the LDH layer, which induces the weakening of the electrophilic character toward C5. Because of the propagation of the shielding effect through the benzene ring, the C1carbon atoms are shifted to down-field values as previously observed for VBS intercalated Zn2Al- and Ca2Al-matrices.35,36 However, the NMR peaks are here rather broad. Such peak broadening may result from a slight distribution in the chemical interaction due to the molecular sequestration, thus resulting in a decrease in the spin relaxation time T2*.
A thermal treatment of 3-DOM Mg2Al–VBS at 493 K induces the complete disappearance of the C1 resonance peak and the concomitant appearance of a broad peak at 40–50 ppm due to the CH and CH2 bonds. These modifications indicate the full polymerization of the monomer between the layers. It is noteworthy that a thermal treatment at lower temperature (473 K) leads to a partial decrease in the intensity of the C1 resonance peak for 3-DOM Mg2Al–VBS only, whereas in the case of Mg2Al–VBS, it is sufficient to result in complete C1 peak extinction (see Fig. S1†). Such a difference of polymerization temperature seems to underline the kinetics barrier for VBS to connect each other in the 3D ordered LDH gap. SEM characterization of the thermally treated sample at 493 K (Fig. 6) shows that neither the in situpolymerization nor the moderate thermal treatment alters the morphology of the 3-DOM compound. A 3-DOM structure is still observed, however, the treatment induces a further shrinkage of the structure and a slight decrease in the macropore size (Table 1).

Carbonization
To access the carbonaceous replica, the last step consists of the carbonization of the polymer under nitrogen and a subsequent dissolution of the inorganic part by acid treatment (Fig. 1). The carbonization temperature is intermediate, being high enough to decompose the LDH structure, but not so high as to promote the crystallization of the oxides and the mixed oxides. The XRD patterns acquired in situ during the carbonization under nitrogen atmosphere (Fig. 6) show the loss of the LDH lamellar structure within the temperature range 773–873 K, giving rise to a nearly flat pattern.Above 873 K, the formation of the well-known oxides MgO and MgAl2O4 is indicated by the appearance of the corresponding diffraction lines. The presence of sulfonate groups does not cause the formation of an unusual by-product under thermal treatment under nitrogen atmosphere, as observed with Zn2Al- and Ca2Al-LDH compositions.36,37 The complete transformation of the VBS-intercalated molecules at 873 K under nitrogen is further confirmed by FTIR spectroscopy, which indicated the absence of characteristic vibration bands (not shown). Acid-leaching treatment is then required to remove the inorganic residual compounds and to access the carbon replica. Recent publications described that through controlled acidic treatment on LDH decomposition products, selective leaching made it possible to prepare porous spinel phases38 or even to produce carbon composites with interesting electrochemical properties.39 In this work, the soaking into 1 M HCl solution ensures complete dissolution of the inorganic residues.
The porosity of the materials was characterized by means of N2 adsorption. The N2 adsorption–desorption isotherm of the starting 3-DOM Mg2Al–VBS hybrid material (Fig. 7a) is typical of LDH mesoporous materials, with a H3 hysteresis loop (IUPAC classification) being present.
 | ||
| Fig. 7 N2 adsorption–desorption isotherms for (1) 3-DOM Mg2Al–VBS and for (2) its corresponding carbon replica. The pore size distribution for the 3-DOM Mg2Al–VBS carbon replica is displayed in the inset. | ||
Due to the highly packed organic molecules between the hydroxylated layers, the hybrid compound displays a low surface area (Table 1). After the calcination and demineralization steps, the N2 isotherm of the resulting carbon shows an adsorption–desorption cycle of type I, and shows a net increase of the gas volume adsorbed at low relative pressures (p/p0). This behavior indicates the generation of microporosity within the material (Fig. 7b).
As previously emphasized,14 the sequestration of organic matter into the LDH structure strongly promotes the microporosity of the resulting carbon. This is also evidenced here, with the preparation of carbons with a much larger surface area than using carbonized VBS or polystyrene sulfonate (PSS) molecules – typically, 1205 m2 g−1 was obtained for 3-DOM carbon and 950 m2 g−1 for C-carbon (see Fig. S2† for the N2 adsorption isotherm), while only 220 and 370 m2 g−1 were measured respectively for VBS and PSS.
In parallel, SEM images were recorded on 3-DOM materials before and after the acid-leaching treatment (Fig. 8). Interestingly, the macroporous structure was observed to be preserved after carbonization of the organic part.
 | ||
| Fig. 8 SEM images at low and high magnification for (a) 3-DOM Mg2Al–VBSp, (b) 3-DOM Mg2Al–VBSp after carbonization under N2 atmosphere at 673 K and (c) after the acid-leaching step. | ||
This result was not obvious, but could be expected since the morphology of the synthesized macroporous phases is stable at high temperature.29 It is to be noted that the residual inorganic part dispersed on carbon matrix was not detectable on the micrograph. The thermal treatment does not induce the formation of extended oxide or mixed oxide individual crystalline particles, the inorganic compounds being rather homogenously embedded within the carbonaceous material. As demonstrated by SEM pictures after acid leaching, the porous architecture is still present and the integrity of the walls is maintained (Fig. 8 and 9).
TEM images of the carbon obtained after inorganic part dissolution are shown in Fig. 9. As for the SEM images, they show the efficiency of the template synthesis for preparing macroporous carbon. The walls of the macroporous structure are composed of 15–30 graphitic layers, whereas the surface of the macropores comprises an association of nanoparticles in a rather open disordered network.
 | ||
| Fig. 9 (a) TEM image and (b) HRTEM image of the carbon replica. | ||
In summary, the opening of the porosity previously ascribed to the dissolution of the mixed oxides14 is not deleterious for the macroporous architecture. Strikingly, the interleaved organic matter transformed into the carbon is able to be stabilized in a 3-DOM structure which remains even after the removal of the initial inorganic stabilizing host. We can assume that this behavior is due to the efficient polymerization of the VBS molecules and their subsequent cross-linking. It should be emphasised that a total shrinkage of the macroporous structure of nearly 50% is observed in comparison with the starting template, concomitantly with a net decrease of the wall thickness (Table 1).
Capacitance properties
At first glance, capacitive performance should be related to specific surface area as far as surface adsorption process is concerned – the so-called electric double-layer capacitance (EDLC). However it has been shown that capacitance is found to reach constant values at large specific area, with no significant increase after 2000 m2 g−1.40 Indeed, it has been shown in a review that ultramicropores and micropores are essential for ion adsorption.41 It was earlier demonstrated that pores smaller than the solvated electrolyte ions are capable of contributing to charge storage, thus resulting in an anomalous increase in carbon capacitance.40,42 However, the contribution of pore sizes less than 1 nm should be efficient in the case of interconnected ultramicropores, since any irregular pore connection will make ionic motion difficult.41 Small mesopores are found to be necessary for rapid transportation of ions to the bulk, and a regular network of small mesopores is often desired, but to the best of our knowledge, few studies have reported on the effect of a porosity hierarchically ordered on three scales. A question arises here: is this multiple degree of porosity promoting the material exposure and then increasing the capacitive performance, or is it deleterious by creating disconnected carbon domains and thus leaving the paths electrically disrupted.To evaluate the influence of the macroporosity on the double-layer charge accumulation, the electrochemical behavior of the 3-DOM carbon, as well as a reference sample with no macroporosity, was studied. For both replicas, cyclic voltammogram curves exhibit a ‘bumpy’ highly distorted rectangular shape, this distortion being even more pronounced at low sweep rates (Fig. 10). This is characteristic of the pseudo-capacitive effect due to the presence of a hetero-element in the carbon material. By varying the potential scan rate ![[scr V, script letter V]](https://www.rsc.org/images/entities/char_e149.gif) , the total charge qT* proportional to the whole active surface is obtained from extrapolation of q* to V → 0 on the 1/q*vs. 1/2 curve (see Fig. S3†) while the outer charge q0* proportional to the outer accessible active surface is extrapolated from the q*vs. −1/2 curve (see Fig. S4†).43–45 Both plots are more or less satisfactorily linear over the entire tested range of . Counterintuitively, 3-DOM carbon replica has poor capacitive performance, lower than our reference. However, the carbon is slightly more exposed in a sense that the inner active surface qi* (obtained using the relation qT* = q0* + qi*) is slightly decreased in relative proportion (88% against 97%). To better understand the reason why performance is not met, electrochemical impedance spectroscopy was studied at both potential limits (Fig. 11). In the high frequency domain (enlarged part of Fig. 11), the intercept of the dashed lines to the Z′-axis corresponds to the equivalent serial resistance (ESR) of the two-electrode capacitor, which is due to the ionic resistivity of the electrolyte present in the porosity of the active material. At both potential limits, the high-frequency response is mostly superimposed for the reference C-carbon, and the ESR value is of about 9 Ω cm−2, indicative of rather easy ion transport, while a large hysteresis is observed for the 3-DOM replica, with an ESR value as large as 115 Ω cm−2 at the higher potential limit. Such a large ESR is characteristic of a strong impediment for the ion to migrate in the vicinity of the electrode material’s double-layers. This may be explained by the carbon network being rather disconnected in the 3-DOM carbon replica (as depicted in Fig. 8 and 9), as well as a small compaction due to the macroscopic open-structure.
, the total charge qT* proportional to the whole active surface is obtained from extrapolation of q* to V → 0 on the 1/q*vs. 1/2 curve (see Fig. S3†) while the outer charge q0* proportional to the outer accessible active surface is extrapolated from the q*vs. −1/2 curve (see Fig. S4†).43–45 Both plots are more or less satisfactorily linear over the entire tested range of . Counterintuitively, 3-DOM carbon replica has poor capacitive performance, lower than our reference. However, the carbon is slightly more exposed in a sense that the inner active surface qi* (obtained using the relation qT* = q0* + qi*) is slightly decreased in relative proportion (88% against 97%). To better understand the reason why performance is not met, electrochemical impedance spectroscopy was studied at both potential limits (Fig. 11). In the high frequency domain (enlarged part of Fig. 11), the intercept of the dashed lines to the Z′-axis corresponds to the equivalent serial resistance (ESR) of the two-electrode capacitor, which is due to the ionic resistivity of the electrolyte present in the porosity of the active material. At both potential limits, the high-frequency response is mostly superimposed for the reference C-carbon, and the ESR value is of about 9 Ω cm−2, indicative of rather easy ion transport, while a large hysteresis is observed for the 3-DOM replica, with an ESR value as large as 115 Ω cm−2 at the higher potential limit. Such a large ESR is characteristic of a strong impediment for the ion to migrate in the vicinity of the electrode material’s double-layers. This may be explained by the carbon network being rather disconnected in the 3-DOM carbon replica (as depicted in Fig. 8 and 9), as well as a small compaction due to the macroscopic open-structure.
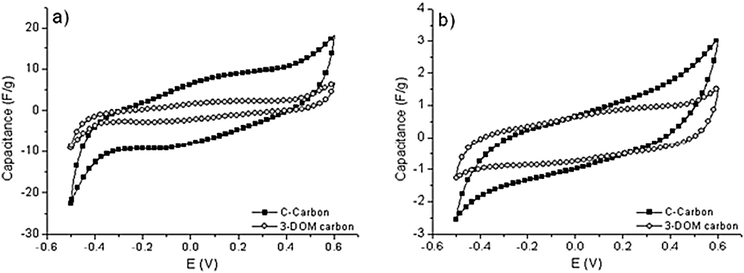 | ||
| Fig. 10 Cyclic voltammogram curves at scan rate of (a) 2 mV s−1 and (b) 100 mV s−1 using 1 M Na2SO3 as the electrolyte for the 3-DOM carbon and C-carbon. | ||
 | ||
| Fig. 11 Complex plane impedance expressed as Z′– jZ′′ Nyquist curves for (a) C-carbon and (b) 3-DOM carbon at imposed potentials. Low impedance domain is enlarged in the inset and the ESR is indicated by a dashed line. | ||
Conclusions
In summary, using a template approach, a 3-DOM LDH hybrid compound intercalated with a VBS monomer has been successfully prepared and further used to produce a hierarchically micro/meso/macroporous carbonaceous replica. While the micropores are characteristic of carbonaceous compounds due to the graphene-plan organization, the mesopores are created during the dissolution of inorganic matter, since the removal of small crystallites acts as porogen agents. The nanostructuration of LDH in inverse opals does not hinder in situpolymerization of the confined VBS, even if a higher temperature is required compared with classical LDH phases. Interestingly, the inverse opal architecture is maintained during the successive steps leading to the chemical modification of the materials. The N2 adsorption–desorption analysis shows that macroporous as-prepared carbon has a surface area of 1205 m2 g−1 containing both micropores and mesopores. In spite of these efforts in designing carbon porosity, low electrochemical performances as EDLC systems in aqueous electrolytes were achieved, indicating that the development of the macroporosity is rather deleterious compared with bi-modal systems, for such applications.References
- G. Centi and S. Perathoner, Eur. J. Inorg. Chem., 2009, 3851 CrossRef CAS.
- A. Huczko, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 365 CrossRef CAS.
- T. Kyotani, Bull. Chem. Soc. Jpn., 2006, 79, 1322 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696 CrossRef CAS.
- H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217 RSC.
- D. Carriazo, F. Pico, M. Gutierrez, F. Rubio, J. M. Rojo and F. del Monte, J. Mater. Chem., 2010, 20, 773 RSC.
- C. O. Ania, V. Khomenko, E. Raymundo-Pinero, J. B. Parra and F. Beguin, Adv. Funct. Mater., 2007, 17, 1828 CrossRef CAS.
- F. O. Gaslain, J. Parmentier, V. P. Valtchev and J. Patarin, Chem. Commun., 2006, 991 RSC.
- T. Kyotani, Z. X. Ma and A. Tomita, Carbon, 2003, 41, 1451 CrossRef CAS.
- Z. X. Ma, T. Kyotani, Z. Liu, O. Terasaki and A. Tomita, Chem. Mater., 2001, 13, 4413 CrossRef CAS.
- P. M. Barata-Rodrigues, T. J. Mays and G. D. Moggridge, Carbon, 2003, 41, 2231–2246 CrossRef CAS.
- M. Darder and E. Ruiz-Hitzky, J. Mater. Chem., 2005, 15, 3913 RSC.
- T. Kyotani, N. Sonobe and A. Tomita, Nature, 1988, 331, 331 CrossRef.
- F. Leroux, E. Raymundo-Pinero, J.-M. Nedelec and F. Beguin, J. Mater. Chem., 2006, 16, 2074 RSC.
- K. Putyera, T. J. Bandosz, J. Jagiello and J. A. Schwarz, Carbon, 1996, 34, 1559 CrossRef CAS.
- T. J. Bandosz, J. Jagiello, K. Putyera and A. J. Schwarz, Chem. Mater., 1996, 8, 2023 CrossRef CAS.
- V. Rives, Layered Doubles Hydroxides: Present and Future, Nova Science Publishers, New York, USA, 2001 Search PubMed.
- A. L. Garcia-Ponce, V. Prévot, B. Casal and E. Ruiz-Hitzky, New J. Chem., 2000, 24, 119 RSC.
- D. Tichit, C. Gérardin, R. Durand and B. Coq, Top. Catal., 2006, 39, 89 CrossRef CAS.
- F. Leroux and J.-P. Besse, Chem. Mater., 2001, 13, 3507 CrossRef CAS.
- R. Ryoo, S. Hoon Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 9.
- A. Stein, F. Li and N. R. Denny, Chem. Mater., 2008, 20, 649 CrossRef CAS.
- L. L. Zhang, S. Li, J. Zhang, P. Guo, J. Zheng and X. S. Zhao, Chem. Mater., 2010, 22, 1195–1202 CrossRef CAS.
- Z. Lei, Y. Zhang, H. Wang, Y. Ke, J. Li, F. Li and J. Xing, J. Mater. Chem., 2001, 11, 1975 RSC.
- S. W. Woo, K. Dokko, K. Sasajima, T. Takei and K. Kanamura, Chem. Commun., 2006, 4099 RSC.
- S.-W. Woo, K. Dokko, H. Nakano and K. Kanamura, J. Mater. Chem., 2008, 18, 1674 RSC.
- Z. Wang, F. Li, N. S. Ergang and A. Stein, Chem. Mater., 2006, 18, 5543 CrossRef CAS.
- E. Géraud, V. Prévot, J. Ghanbaja and F. Leroux, Chem. Mater., 2006, 18, 238 CrossRef CAS.
- E. Geraud, S. Rafqah, M. Sarakha, C. Forano, V. Prevot and F. Leroux, Chem. Mater., 2008, 20, 1116 CrossRef CAS.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738 CrossRef CAS.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS.
- E. Géraud, V. Prévot and F. Leroux, J. Phys. Chem. Solids, 2006, 67, 903 CrossRef CAS.
- T. Stanimirova and G. Kirov, Appl. Clay Sci., 2003, 22, 295 CrossRef CAS.
- E. M. Moujahid, J.-P. Besse and F. Leroux, J. Mater. Chem., 2002, 12, 3324 RSC.
- L. Vieille, C. Taviot-Gueho, J.-P. Besse and F. Leroux, Chem. Mater., 2003, 15, 4369 CrossRef CAS.
- F. Leroux and M. Dubois, J. Mater. Chem., 2006, 16, 4510 RSC.
- L. Zou, F. Li, X. Xiang, D. G. Evans and X. Duan, Chem. Mater., 2006, 18, 5852 CrossRef CAS.
- T. Stimpfling and F. Leroux, Chem. Mater., 2010, 22, 974 CrossRef CAS.
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowsski and F. Beguin, Carbon, 2006, 44, 2498 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774 RSC.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760 CrossRef CAS.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263 CrossRef CAS.
- C. P. De Pauli and S. Trasatti, J. Electroanal. Chem., 1995, 396, 161 CrossRef CAS.
- M. Toupin, T. Brousse and D. Belanger, Chem. Mater., 2002, 14, 3946 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR spectra, N2 adsorption–desorption isotherm, variation of the voltammetric charge. See DOI: 10.1039/c0nj00630k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |