A self-assembled bis(pyrrolo)tetrathiafulvalene-based redox active square†
Jean-Yves
Balandier
,
Marcos
Chas
,
Sébastien
Goeb
,
Paul I.
Dron
,
David
Rondeau
,
Ahmed
Belyasmine
,
Nuria
Gallego
and
Marc
Sallé
*
Université d'Angers, CNRS UMR 6200, MOLTECH-Anjou, 2 Bd Lavoisier, 49045, Angers Cedex, France. E-mail: marc.salle@univ-angers.fr; Fax: +33 241735405; Tel: +33 241735439
First published on 11th October 2010
Abstract
The synthesis and full characterization of a redox-active π-donating molecular square is described, through coordination-driven self-assembly of a new key bis(pyrrolo)TTF building block with Pt(II) salts; the electrochemical behaviour of the square is consistent with noninteracting bis(pyrrolo)TTF units.
Introduction
The coordination-driven self-assembly methodology has proven to be of particular interest for preparing molecular polygons and polyhedrons of predetermined shapes,1 such systems being otherwise very challenging to synthesize through classical covalent multistep synthesis. These supramolecular assemblies generally call for a combination of rigid di-, tri- or tetra pyridyl substituted bridging ligands and complementary metallic salts of defined geometries (e.g. square planar Pt(II) and Pd(II) salts). Beside the synthetic challenge of preparing large size host architectures, they offer unique opportunities for new applications which are directly correlated to the nature of their two constituting partners (ligand and metal complexes), such as molecular recognition.1 In this context, some examples of supramolecular polygons involving redox-active units have been described, with electroactivity located on the corner (i.e. the metal itself or an organometallic ligand (e.g.ferrocene) directly bound to the metal).1,2Ferrocene unit can also be found as a pendant group on the bridging ligand between two metallic corners.3 Noteworthy, only few examples of polygons involving side walls that are themselves redox-active are described,4 and in this case, they involve reducible ligands such as 4,4′-bipyridine or perylene diimide. This spatial organization allows a control over the charge state of the cavity. However, to the best of our knowledge, functional metallosupramolecular squares built from highly π-donating side-walls are unknown to date, and would constitute a challenging reciprocal complement to previous π-electron accepting supramolecular or covalent macrocycles among which is found the famous cyclophane cyclobis(paraquat-p-phenylene) (CBPQT4+),5a,b extensively used in donor–acceptor [2]-rotaxane chemistry.5c The π-donating ability of tetrathiafulvalene (TTF) derivatives is well-established, and has projected this unit as a key redox building block in various molecular and supramolecular switchable systems.6 In particular, TTF derivatives are very easily oxidized according to two reversible one e− steps.We propose here the preparation of a redox-active π-donating supramolecular square by self-assembling of a new TTF-based ligand with Pt(II) phosphine corners. Several TTF derivatives bearing two pyridyl coordinating moieties are described.7 These systems have not been selected to our purpose since they exist as two Z/E isomers (Scheme 1), known to be very difficult to separate and which undergo a Z/Eisomerization after oxidation or protonation.8 We have instead focused our interest on the non-isomerizable derivative 1 as a key redox-active ligand. This system involves the bis(pyrrolo)TTF skeleton (BPTTF)9 as a basic framework, a unit which occupies a growing place among electroactive supramolecular architectures.6a,10
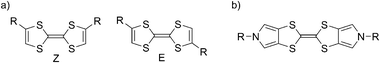 | ||
| Scheme 1 (a) The two isomeric Z and E forms in bis-substituted TTF; (b) N,N′-bis-substituted BPTTF. | ||
Results and discussion
We recently described the synthesis of the key thione derivative 311 (Scheme 2), starting from 4-formyl-5-(diethoxymethyl)-1,3-dithiole-2-thione12 and involving a three step procedure.9b The target bis-pyridyl BPTTF derivative 1 is obtained by self-coupling of thione 3 in refluxing trimethylphosphite. DFT calculations (Gaussian® 2003, Becke3LYP (B3LYP)) have been carried out on compound 1 (Fig. 1). The electronic density in the HOMO of 1 is located on the central donating skeleton and on the lateral pyrrole motifs, illustrating the strong π-delocalization in BPTTF derivatives compared to the parent TTF framework; the LUMO is essentially located on both pyridyl and pyrrole heterocycles. The corresponding energies are very similar to that of unsubstituted BPTTF, anticipating a comparably high π-donating ability.10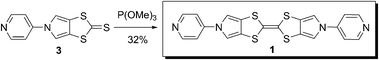 | ||
| Scheme 2 Synthesis of bis(pyridyl)-BPTTF ligand 1. | ||
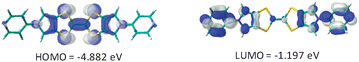 | ||
| Fig. 1 Frontier orbitals of compound 1. | ||
Reaction of 1 with a cis-blocked ethylene diamine Pd(II) complex (enPd(NO3)2) in DMSO-d6 could be monitored by 1H NMR. The initial spectrum, which corresponds to a complex mixture, progressively converges to a simple set of five signals after four hours at rt (Fig. S2, ESI†), as expected from the formation of a polygon symmetric structure. Attempts to isolate the complex from the NMR solution failed. On the other hand, the use of the more kinetically inert Pt(II) metal complex (enPt(NO3)2) only leads to a mixture of complexes even after heating at 100 °C for two weeks in DMSO-d6. On the contrary, the reaction of 1 with an equimolar amount of cis-blocked complex dpppPt(OTf)2 in methylene chloride leads to the rapid dissolution of the ligand, and subsequent precipitation of a yellow powder corresponding to the target complex 2, isolated in a 31% yield after filtration (Scheme 3). Molecular square 2 is soluble in different organic solvents (acetone, acetonitrile, nitromethane, DMF, DMSO), and could be characterized through various techniques. Multinuclear NMR (1H, 31P, 19F) analyses in acetone-d6 are illustrative of the formation of a discrete supramolecule (Fig. 2). Only two AA′XX′ signals are observed for pyridyl protons (Hα = 8.75 ppm and Hβ = 7.28 ppm) in accordance with pyridine units coordinated to dpppPt(OTf)2.13Metal binding is furthermore confirmed by a downfield shift of the CH2P signal in 2 relative to dpppPt(OTf)2 complex (Table S1, ESI†). The 19F spectrum as well as the 31P NMR (−13.4 ppm, accompanied by 195Pt–31P satellites) exhibits a single signal (−79.2 ppm), illustrating the occurrence of a single discrete structure. Diffusion-ordered spectroscopy (DOSY) NMR has become a valuable tool for investigating large molecules.14 A DOSY experiment performed on 2 supports the occurrence of a single polygon species, as illustrated by the alignment of a single set of diffusion coefficient value in the spectrum (Fig. 3, left). NMR data do provide an insight into the symmetry of the polygon, but are not enough to extract polygon size information. The use of mass spectrometry to characterize metal-assembled polygons has been demonstrated,15 in particular through the use of the high resolving power of a Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer. In our case, an experimental isotopic pattern centered on the m/z 1640 peak is observed in the ESI-FTICR mass spectrum of 2 (Fig. 3, right and Table S2, ESI†). The comparison with a theoretical isotopic distribution calculated for a tricharged species (Δm = 0.33 u) confirms the transfer into the gas-phase of the molecular square structure [(1–Ptdppp)4·5OTf]3+. Other charged species are also observed in the ESI mass spectra of 2 such as [1–Ptdppp·OTf]+ at m/z 1192 and [(1–Ptdppp)2·OTf]3+at m/z 745. They correspond to a quarter and a half of a square, respectively (Table S2, ESI†), and their presence can be attributed either to a formation in solution prior to the electrospray process or to fragmentations due to in-source collision induced dissociations.
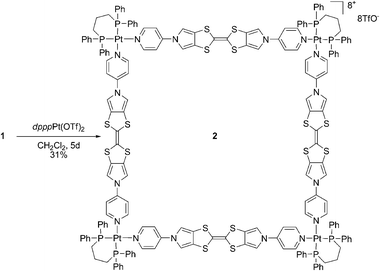 | ||
| Scheme 3 Synthesis of square 2. | ||
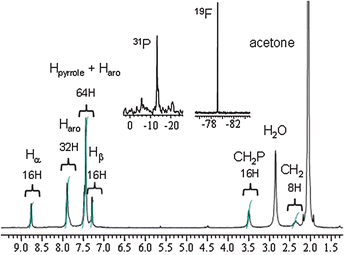 | ||
| Fig. 2 1H, 31P and 19F NMR signals of 2 (acetone-d6). | ||
![Left: DOSY experiment of 2 (acetone-d6), 500 MHz, 298 K; right: ESI-FT-ICR mass spectrum of 2 in acetone. Comparison of the experimental isotopic pattern of the triply charged ion [(1–Ptdppp)4·5OTf]3+ (up) with the calculated isotope pattern (bottom).](/image/article/2011/NJ/c0nj00545b/c0nj00545b-f3.gif) | ||
| Fig. 3 Left: DOSY experiment of 2 (acetone-d6), 500 MHz, 298 K; right: ESI-FT-ICR mass spectrum of 2 in acetone. Comparison of the experimental isotopic pattern of the triply charged ion [(1–Ptdppp)4·5OTf]3+ (up) with the calculated isotope pattern (bottom). | ||
Cyclic voltammetry of 216 was carried out in CH2Cl2/CH3CN (1/1 v/v) (Bu4NPF6 0.1 M), and allows observation of two redox processes corresponding to the reversible oxidation of the four BPTTF moieties (E1ox = 630 mV et E2ox = 920 mV) which behave independently (Fig. 4). In addition, the Pt(II) corner appears electrochemically inert in this window of potentials.4b Square 2 presents a good stability under its different oxidation states, as shown by the shape of the CV which remains unchanged upon recurrent scanning of the potentials between 0.3 and 1.1 V vs.AgCl/Ag, a behavior which is identical to previous observations made on the half-molecule, built from the association of two redox BPTTF entities and one Ptdppp complex.11 Thin Layer Cyclic Voltammetry (TLCV)17 experiments performed on this model system and using ferrocene as an internal reference, have shown that each BPTTF unit behaves independently, displaying two successive 1e− reversible processes for each BPTTF unit upon oxidation.11 Unfortunately, because of adsorption phenomena, TLCV studies failed in the case of compound 2, and did not allow to probe coulometric data. Considering the similar electrochemical CV responses between 2 and the model half-molecule, it is nevertheless reasonable to anticipate that square 2 integrating four BPTTF units, behaves similarly, with each of the four BPTTF units capable to reversibly generate a dicationic species, thus providing a reversible control over the charge state of the cavity. This renders this self-assembled square structure particularly attractive as a very easily oxidisable π-donating macrocyclic system complementary to the extensively studied reducible ones generally based on the bipyridinium unit.
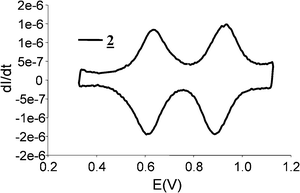 | ||
| Fig. 4 Deconvoluted cyclic voltammogram of 2 (5 × 10−4 M), NBu4PF6 (10−1 M), v = 100 mV s−1, vs.AgCl/Ag, CH3CN/CH2Cl2 (1/1). | ||
Several electron-rich receptors incorporating TTF units or derivatives (extended-TTF, BPTTF)6e,11,18 have been shown to present good binding ability for electrodeficient C60. The binding affinity of square 2 for C60 was studied by UV-visible titration. Increasing amounts of C60 were added to a solution of 2 in dichloromethane (Fig. S3, ESI†). The titration curve shows the expected decrease of the absorption band at 395 nm corresponding to disappearing of receptor 2, but did not allow observation of the complex, expected at ca. 600 nm; instead the solution becomes turbid, presumably due to precipitation.
In order to evaluate the cavity size of such polygon, DFT calculations have been performed on the Pd square (1/enPd(NO3)2) (Fig. S4, ESI†). From this study, the metal centers show a square planar geometry, in which bond distances and angles are in the usual range. Plane to plane distances of ca. 22 Å were found between facing BPTTF units, and a value of 32.5 Å for the diagonal of the square. This corresponds to a cavity size which is roughly double compared to the parent square built from a 4,4′-bipyridine bridging ligand,13 which makes this system one of the largest squares assembled to date, and which justifies the lack of visible interaction between redox units during CV analyses.
Conclusions
A redox-active molecular square which incorporates the highly π-donating bis(pyrrolo)-TTF unit has been synthesized. Besides to constitute the first synthetic response to a TTF-square target structure suggested by Fabre a decade ago in conclusion of a review,19 this system allows a control over the charge state of the corresponding cavity upon reversible oxidation. As an easily oxidisable macrocycle, it constitutes therefore a promising reciprocal complement to the extensively used reducible π-electron deficient supramolecular or covalent macrocyclic analogues.Association studies of this metalla-ring with various π-accepting species are underway.
Experimental
Bis-(N-(4-pyridyl)pyrrolo[3,4-d])tetrathiafulvalene 1
Dithiole thione 8 (267 mg, 1.07 mmol) is dissolved in freshly distilled trimethylphosphite (20 mL). The reaction mixture is refluxed for 4 h. After cooling, methanol (30 mL) is added and the resulting solid is separated by filtration (frit No 4), and rinsed successively with methanol, dichloromethane, acetone, and diethyl ether, to produce 1 as an orange powder (61 mg, 0.14 mmol). Yield 26%; mp > 260 °C; C20H12N4S4: MALDI-TOF: M+˙: 435.99; IR (KBr): 1645, 1593, 1511, 1316 cm−1. Though insoluble in common organic solvents, a 1H NMR analysis could be carried out in DMSO-d6, by adding BF3/Et2O to a suspension of 1 in DMSO-d6 (Fig. S1, ESI†), producing therefore in situ a more soluble bis(N(pyridyl)-BF3) derivative. 1H NMR (DMSO-d6): 8.83 (d, 4 H, J = 5.0 Hz, Hα), 8.09 (d, 4 H, J = 5.0 Hz, Hβ), 7.91 (s, 4 H, Hpyrrole).Square [(1–Ptdppp)4]8+·8OTf−2
To a solution of dpppPt(OTf)2 (21.9 mg, 24 μmol) in CH2Cl2 (12 mL) is added ligand 1 (9.6 mg, 22 μmol). The reaction mixture is stirred for 5 days at rt in the darkness. The resulting solid formed along the reaction is filtered off (frit No 4) and rinsed with CH2Cl2 (10 mL), to afford a yellow-orange powder (9.3 mg, 1.7 μmol). Yield 31%; mp > 260 °C; 1H NMR (acetone-d6): 8.75 (d, 16 H, J = 5.16 Hz, Hα), 7.89 (m, 36 H, Haro), 7.44 (m, 60 H, Haro + Hpyrrole), 7.28 (d, 16 H, J = 5.16 Hz, Hβ), 3.49 (m, 16 H, CH2P), 2.35 (m, 8 H, CH2); 31P NMR (acetone-d6): −13.4 (JPt–P = 3049 Hz); 19F NMR (acetone-d6): −79.2; C193H152F15N16O15P8Pt4S21: FT-ICR: [1–3OTf]3+ (calc.): 1640.4075; [1–3OTf]3+ (tr.): 1640.4002.Acknowledgements
The authors gratefully acknowledge Dr C. Afonso for his help during ESI-FTICR analyses (SM3P platform), Pr P. Richomme and Dr E. Levillain for their assistance in DOSY and TLCV analyses, respectively, and Dr D. Amabilino for fruitful discussions. This work has been financially supported by ANR-PNANO (ANR-07-NANO-030-01).Notes and references
- For recent reviews, see: (a) S. S. Li, B. H. Northrop, Q. H. Yuan, L. J. Wan and P. J. Stang, Acc. Chem. Res., 2009, 42, 249–259 CrossRef CAS; (b) P. J. Stang, J. Org. Chem., 2009, 74, 2–20 CrossRef CAS; (c) B. H. Northrop, D. Chercka and P. J. Stang, Tetrahedron, 2008, 64, 11495–11503 CrossRef CAS; (d) B. H. Northrop, H. B. Yang and P. J. Stang, Chem. Commun., 2008, 5896–5908 RSC; (e) E. Zangrando, M. Casanova and E. Alessio, Chem. Rev., 2008, 108, 4979–5013 CrossRef CAS; (f) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS; (g) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS; (h) A. Kaiser and P. Bauerle, Top. Curr. Chem., 2005, 249, 127–201 CAS; (i) F. Würthner, C. C. You and C. R. Saha-Möller, Chem. Soc. Rev., 2004, 33, 133–146 RSC.
- (a) P. J. Stang, B. Olenyuk, J. Fan and A. M. Arif, Organometallics, 1996, 15, 904–908 CrossRef CAS; (b) S. S. Sun and A. J. Lees, Inorg. Chem., 2001, 40, 3154 CrossRef CAS.
- (a) H. B. Yang, K. Ghosh, Y. Zhao, B. H. Northrop, M. M. Lyndon, D. C. Muddiman, H. S. White and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 839–841 CrossRef CAS; (b) K. Ghosh, Y. Zhao, H. B. Yang, B. H. Northrop, H. S. White and P. J. Stang, J. Org. Chem., 2008, 73, 8553–8557 CrossRef CAS; (c) C. C. You and F. Würthner, J. Am. Chem. Soc., 2003, 125, 9716–9725 CrossRef CAS.
- (a) W. Kaim, B. Schwederski, A. Dogan, J. Fiedler, C. J. Kuehl and P. J. Stang, Inorg. Chem., 2002, 41, 4025–4028 CrossRef CAS; (b) F. Würthner, A. Sautter, D. Schmid and P. J. A. Weber, Chem.–Eur. J., 2001, 7, 894–902 CrossRef CAS.
- (a) B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547 CrossRef; (b) The dynamic character of the covalent bond in CBPQT4+ ring with iodide has been recently demonstrated: O. S. Miljanic and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12966 Search PubMed; (c) N. N. P. Moonen, A. H. Flood, J. M. Fernández and J. F. Stoddart, Top. Curr. Chem., 2005, 262, 99–132 CAS.
- For reviews, see: (a) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC; (b) N. Martín and J.-L. Segura, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS; (c) M. R. Bryce, J. Mater. Chem., 2000, 10, 589–598 RSC; (d) M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153–164 RSC; (e) M. Hardouin-Lerouge, P. Hudhomme and M. Sallé, Chem. Soc. Rev., 2010 10.1039/b915145c.
- (a) D. Lorcy, N. Bellec, M. Fourmigue and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS; (b) Y. Geng, X.-J. Wang, B. Chen, H. Xue, Y.-P. Zhao, S. Lee, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2009, 15, 5124–5129 CrossRef CAS; (c) J.-Y. Balandier, A. Belyasmine and M. Sallé, Eur. J. Org. Chem., 2008, 269–276 CrossRef CAS; (d) H. Xue, X. J. Tang, L. Z. Wu, L. P. Zhang and C. H. Tung, J. Org. Chem., 2005, 70, 9727 CrossRef CAS.
- (a) A. Souizi, A. Robert, P. Batail and L. Ouahab, J. Org. Chem., 1987, 52, 1610 CrossRef CAS; (b) F. Le Derf, M. Mazari, N. Mercier, E. Levillain, G. Trippe, A. Riou, P. Richomme, J. Becher, J. Garín, J. Orduna, N. Gallego-Planas, A. Gorgues and M. Salle, Chem.–Eur. J., 2001, 7, 447–455 CrossRef CAS; (c) S. Nygaard, S. W. Hansen, J. C. Huffman, F. Jensen, A. H. Flood and J. O. Jeppesen, J. Am. Chem. Soc., 2007, 129, 7354–7363 CrossRef CAS.
- (a) J. O. Jeppesen, K. Takimiya, F. Jensen, T. Brimert, K. Nielsen, N. Thorup and J. Becher, J. Org. Chem., 2000, 65, 5794–5805 CrossRef; (b) J.-Y. Balandier, A. Belyasmine and M. Sallé, Synthesis, 2006, 2815–2817 CAS.
- J. O. Jeppesen and J. Becher, Eur. J. Org. Chem., 2003, 3245–3266 CrossRef CAS.
- J. Y. Balandier, M. Chas, P. Dron, S. Goeb, D. Canevet, A. Belyasmine, M. Allain and M. Sallé, J. Org. Chem., 2010, 75, 1589–1599 CrossRef CAS.
- M. Sallé, A. Gorgues, M. Jubault, K. Boubekeur and P. Batail, Tetrahedron, 1992, 48, 3081–3090 CrossRef CAS.
- P. J. Stang and D. H. Cao, J. Am. Chem. Soc., 1994, 116, 4981 CrossRef CAS.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS.
- C. A. Schalley, T. Müller, P. Linnartz, M. Witt, M. Schäfer and A. Lützen, Chem.–Eur. J., 2002, 8, 3538–3551 CrossRef CAS.
- Comparison with 1 could not be done because of its insolubility.
- R. Carlier, P. Frère, M. Sallé, J. Roncali, M. Jubault, A. Tallec and A. Gorgues, Adv. Mater., 1993, 5, 445 CrossRef CAS.
- (a) H. Isla, M. Gallego, E. M. Pérez, R. Viruela, E. Ortí and N. Martín, J. Am. Chem. Soc., 2010, 132, 1772–1773 CrossRef CAS; (b) S. S. Gayathri, M. Wielopolski, E. M. Pérez, G. Fernández, L. Sánchez, R. Viruela, E. Ortí, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2009, 48, 815 CrossRef CAS; (c) E. M. Pérez, A. L. Capodilupo, G. Fernández, L. Sánchez, P. M. Viruela, R. Viruela, E. Ortí, M. Bietti and N. Martín, Chem. Commun., 2008, 4567–4569 RSC; (d) E. M. Pérez, L. Sánchez, G. Fernández and N. Martín, J. Am. Chem. Soc., 2006, 128, 7172–7173 CrossRef CAS; (e) E. M. Pérez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC; (f) K. A. Nielsen, G. H. Sarova, L. M. Martin-Gomis, F. Fernández-Lázaro, P. C. Stein, L. Sanguinet, E. Levillain, J. L. Sessler, D. M. Guldi, A. A. Sastre-Santos and J. O. Jeppesen, J. Am. Chem. Soc., 2008, 130, 460–462 CrossRef CAS; (g) K. A. Nielsen, W.-S. Cho, G. H. Sarova, B. M. Petersen, A. D. Bond, J. Becher, F. Jensen, D. M. Guldi, J. L. Sessler and J. O. Jeppesen, Angew. Chem., Int. Ed., 2006, 45, 6848–6853 CrossRef CAS.
- J. M. Fabre, J. Phys. IV, 2000, 10, 3–19 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR, FT-ICR data as well as molecular modeling data. See DOI: 10.1039/c0nj00545b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |