New molecular design of donor-π-acceptor dyes for dye-sensitized solar cells: control of molecular orientation and arrangement on TiO2 surface
Yousuke
Ooyama
,
Yoshihito
Shimada
,
Shogo
Inoue
,
Tomoya
Nagano
,
Youhei
Fujikawa
,
Kenji
Komaguchi
,
Ichiro
Imae
and
Yutaka
Harima
*
Department of Applied Chemistry, Graduate School of Engineering, Hiroshima University, Higashi-, H, iroshima 739-8527, Japan. E-mail: harima@mls.ias.hiroshima-u.ac.jp; Fax: +81 82-424-5494
First published on 11th October 2010
Abstract
A series of benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type fluorescent dyes OH1, OH2, OH4, OH7, and OH17 with carboxyl groups on different positions of a chromophore skeleton are synthesized and applied to dye-sensitized solar cells (DSSCs) as a new class of donor-π-acceptor (D-π-A) photosensitizers. In the dye OH1, a carboxyl group acts as not only the anchoring group for attachment on TiO2 surface but also the electron acceptor. For OH2, OH4, and OH7, on the other hand, a carboxyl group is an anchoring group, but the electron acceptor is not a carboxyl group but a cyano group. The dye OH17 has two carboxyl groups, which are located at the same positions as those of OH1 and OH7. The absorption and fluorescence spectra and cyclic voltammograms of these fluorescent dyes resemble very well, showing a negligible influence of the position of carboxyl group on photophysical and electrochemical properties of these dyes. When these dyes are used in DSSCs, however, their photovoltaic performances differ considerably. In order to elucidate the difference in the DSSC performance among the five dyes, kinetics of the electron injection from the conduction band of TiO2 to dye cation and to I3− ions in solution were studied by employing the transient absorption spectroscopy and the transient photovoltage technique, respectively. It is found that the charge recombination rate for OH2 is similar to that of OH1 and is much slower than those of OH4 and OH17, consistent with the high short-circuit photocurrent densities for OH1 and OH2. On the basis of the MO calculations (AM1 and INDO/S) and the charge recombination kinetics, the differences of the photovoltaic performances among the five dyes are discussed from the viewpoint of configuration of the dyes adsorbed on TiO2 surface, and a new molecular design for D-π-A dye sensitizers based on a control of molecular orientation and arrangement of dye adsorbed on TiO2 surface is proposed.
Introduction
Since Grätzel and co-workers reported high solar cell performances for dye-sensitized solar cells (DSSCs) based on a Ru-complex dye in 1991,1 DSSCs have received considerable attention because of high incident solar light-to-electricity conversion efficiency and low cost of production.2–13 A diagrammatic illustration of the construction and the operational principle of typical DSSCs are shown in Fig. 1. To begin with, the dye sensitizer absorbs a photon (sun light) to generate a photoexcited-state of dye (S*) (1). Next, the photoexcited dye (S*) injects an electron to the conduction band (CB) (−0.5 V vs.NHE) of TiO2 (2). The resultant oxidized dye (dye cation) is subsequently reduced back to its original neutral state by electron donation from the iodide ion I− in the redox solution; this process is usually called dye regeneration or re-reduction (3). The injected electrons in CB move through the network of interconnected TiO2 nanoparticles to arrive at the transparent conducting oxide (FTO) and then through the external circuit to the counter electrode (Pt-coated glass). The iodide ion I− is regenerated by the reduction of triiodide ion I3− at the counter electrode through the donation of electrons from the external circuit (4) and then the electron flow cycle is completed. During this cycle, however, there are undesirable side processes: the electrons injected to the CB of TiO2 may reduce either oxidized dye (recombination) (5) or I3− in solution (dark current) (6). These two processes (5) and (6) lower the short-circuit photocurrent density (Jsc) and the open-circuit photovoltage (Voc) of DSSCs, respectively. The recombination process (5) competes with the re-reduction of the oxidized dyes by I− (3). To generate greater Jsc and Voc, therefore, the electron injection (2) and the regeneration (3) processes must be much faster than those of the recombination (5) and dark current (6).2–7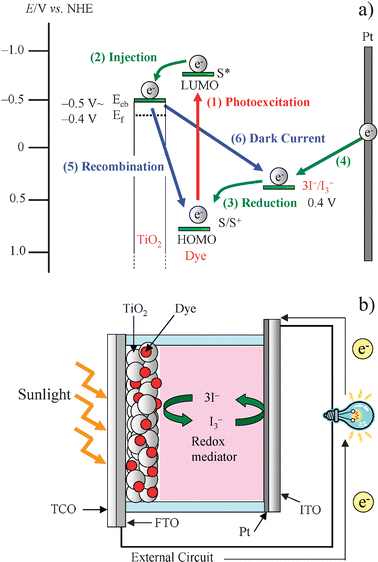 | ||
| Fig. 1 Diagrammatic illustration of (a) construction and (b) operational principle of DSSCs. | ||
Recently, much research has focused on the development of new metal-free organic dyes. In particular, donor–acceptor π-conjugated (D-π-A) dyes, with both the electron-donating (D) and electron-accepting (A) groups linked by a π-conjugated bridge exhibiting broad and intense absorption spectra, are proposed as one of the most promising organic dye sensitizers.5–13 Most of the D-π-A photosensitizers have dialkylamine or diphenylamine moiety as electron donor, and carboxylic acid, cyanoacrylic acid or rhodanine-3-acetic acid moiety which acts as electron acceptor as well as anchoring group for attachment on TiO2 surface. The carboxyl group enables a good electron communication between the dye and TiO2 by forming a strong ester linkage with TiO2 surface. The spectral features of the D-π-A dyes are associated with the intramolecular charge transfer (ICT) excitation from the donor to acceptor moiety of the dye, leading to efficient electron transfer from the excited dye through the acceptor moiety (carboxyl group) into the CB of TiO2.
In our previous study, a series of novel benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type fluorescent dyes OH1–4 with carboxyl groups on different positions of a chromophore skeleton have been designed and synthesized as a new class of D-π-A photosensitizers (Scheme 1).14 In OH1, a carboxyl group acts as not only the anchoring group for attaching on TiO2 surface but also the electron acceptor. For OH2, OH3, and OH4, on the other hand, a carboxyl group is an anchoring group, but the electron acceptor is not a carboxyl group but a cyano group. The photovoltaic performance of OH2 having a non-conjugated linkage between a carboxyl group and a chromophore is similar to that of OH1 and is higher than those of OH3 and OH4. In OH1, electrons can be injected efficiently from the carboxyl group to the CB of a TiO2 electrode through the ester linkage with the TiO2 surface. On the other hand, from the molecular structure of OH2, it was suggested that the cyano group acting as electron acceptor is located close to the TiO2 surface. Consequently, dye OH2 can inject efficiently electrons from the cyano group to the CB of a TiO2 electrode. In order to provide a further confirmation for this view, dyes OH5–7 with different lengths of non-conjugated alkyl chains containing a carboxyl group at the end position have been designed and synthesized (Scheme 1).15 It is found that in spite of the lengths of the alkyl chains, due to flexibility of alkyl chain, the cyano group of the dyes is located near the TiO2 surface and thus a good electron communication between the dyes and the TiO2 surface is established. It is concluded that a carboxyl group of D-π-A sensitizer is necessary not as an electron acceptor, but only as an anchoring group for attachment on the TiO2 surface. This implies that the essential point for developing new and efficient D-π-A sensitizers for DSSCs is to design dye molecules capable of forming a strong interaction between the electron acceptor moiety of sensitizers and the TiO2 surface.14–17
![A series of benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type fluorescent dyes.](/image/article/2011/NJ/c0nj00585a/c0nj00585a-s1.gif) | ||
| Scheme 1 A series of benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type fluorescent dyes. | ||
The aim of this paper is to investigate the effect of the charge recombination (5) and the dark current (6) on the photovoltaic performances of benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type fluorescent dyes OH1, OH2, OH4, OH7, and OH17 by using the transient absorption spectroscopy and the transient photovoltage technique. On the basis of the MO calculations (AM1 and INDO/S) and the kinetic information on the undesirable processes (5) and (6), the differences of photovoltaic performances among the five dyes are discussed by taking account of possible configurations of the dyes adsorbed on TiO2 surface, and a new molecular design for D-π-A dye sensitizers based on a control of molecular orientation and arrangement of dye adsorbed on TiO2 surface is proposed.
Results
The optical and electrochemical data, the HOMO and LUMO energy levels, and the DSSC performance parameters of OH1, OH2, OH4, OH7, and OH17 are summarized in Table 1. The absorption (λmaxabs = 416–431 nm, ε = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100–26100 dm3 mol−1 cm−1) and fluorescence spectra (λmaxfl = 525–541 nm, Φ = 0.95–0.99) in 1,4-dioxane and cyclic voltammograms (Eox = 0.28–0.34 V) of these dyes resemble very well, showing that the position of carboxyl group has a negligible influence on the photophysical and electrochemical properties of these dyes. On the other hand, the absorbance (0.8–0.9) and absorption peak wavelengths (410–430 nm) of adsorbed dyes on TiO2 films are very similar to one another. The HOMO and LUMO energy levels of these dyes lie in the range of 0.86 to 0.91 V and of −1.63 to −1.75 V, respectively, indicating that all these dyes have similar HOMO and LUMO energy levels. Evidently, the LUMO energy levels for the five dyes are higher than the energy level of the CB of TiO2 (−0.5 V), so that these dyes can efficiently inject electrons to the TiO2 electrode. Furthermore, it can be presumed from the five dyes having similar HOMO energy levels that the rate of the dye regeneration by electron donation from I− in solution is similar to one another.
100–26100 dm3 mol−1 cm−1) and fluorescence spectra (λmaxfl = 525–541 nm, Φ = 0.95–0.99) in 1,4-dioxane and cyclic voltammograms (Eox = 0.28–0.34 V) of these dyes resemble very well, showing that the position of carboxyl group has a negligible influence on the photophysical and electrochemical properties of these dyes. On the other hand, the absorbance (0.8–0.9) and absorption peak wavelengths (410–430 nm) of adsorbed dyes on TiO2 films are very similar to one another. The HOMO and LUMO energy levels of these dyes lie in the range of 0.86 to 0.91 V and of −1.63 to −1.75 V, respectively, indicating that all these dyes have similar HOMO and LUMO energy levels. Evidently, the LUMO energy levels for the five dyes are higher than the energy level of the CB of TiO2 (−0.5 V), so that these dyes can efficiently inject electrons to the TiO2 electrode. Furthermore, it can be presumed from the five dyes having similar HOMO energy levels that the rate of the dye regeneration by electron donation from I− in solution is similar to one another.
| Dye | λ abs/nm (ε/M−1cm−1)a | λ em/nm (Φf)b | E ox c/V | HOMOd/V | LUMOd/V | Molecules cm−2e | J sc f/mA cm−2 | V oc f/mV | ff f | η f (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a In 1,4-dioxane. b Fluorescence quantum yields (Φ) were determined by using a calibrated integrating sphere system (λex = 325 nm). c Oxidation potentials (Eox) were recorded in MeCN/Et4NClO4 (0.1 M) solution with a three-electrode system consisting of Ag/Ag+ as a reference electrode, Pt plate as a working electrode, and Pt wire as a counter electrode. d Vs. normal hydrogen electrode (NHE). e Adsorption amount per unit area of TiO2 film. f The photocurrent–voltage characteristics were measured under a simulated solar light (AM 1.5, 61 mW cm−2). | ||||||||||
| OH1 | 416 (25700) |
525 (0.97) | 0.28 | 0.87 | −1.71 | 6.8 × 1016 | 2.12 | 508 | 0.57 | 1.00 |
| OH2 | 425 (24400) |
537 (0.99) | 0.34 | 0.91 | −1.64 | 5.5 × 1016 | 2.01 | 548 | 0.56 | 1.00 |
| OH4 | 424 (22200) |
541 (0.97) | 0.33 | 0.91 | −1.63 | 6.6 × 1016 | 1.50 | 470 | 0.58 | 0.67 |
| OH7 | 431 (26100) |
533 (0.95) | 0.30 | 0.86 | −1.75 | 7.4 × 1016 | 1.91 | 520 | 0.57 | 0.90 |
| OH17 | 417 (21100) |
522 (0.97) | 0.31 | 0.89 | −1.65 | 6.5 × 1016 | 1.30 | 488 | 0.59 | 0.61 |
The MO calculations (AM1 and INDO/S) showed that the absorption bands of these dyes were mainly assigned to the transition from HOMO to LUMO, where HOMOs were mostly localized on the 3-dibutylamino-benzofuro[2,3-c]oxazolo[4,5-a]carbazole moiety for all these dyes, and LUMOs were localized mostly on the carboxyphenyl moiety for OH1 and OH17 and the cyanophenyl moiety for OH2, OH4, and OH7, suggesting a strong ICT nature upon illumination (Fig. 2).
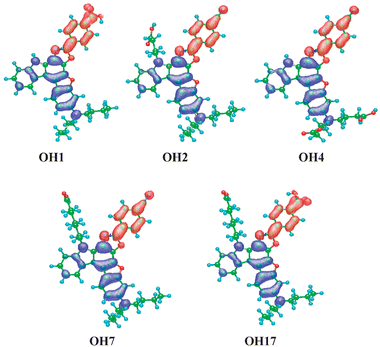 | ||
| Fig. 2 Calculated electron density changes accompanying the first electronic excitation of OH1, OH2, OH4, OH7, and OH17. The blue and red lobes signify decrease and increase in electron density accompanying the electronic transition, respectively. Their areas indicate the magnitude of the electron density change (light blue, green, blue and red balls correspond to hydrogen, carbon, nitrogen, and oxygen atoms, respectively). | ||
When the performances of DSSCs fabricated using these dyes as sensitizers were examined, however, large differences in the incident photon-to-current conversion efficiency (IPCE) spectra and the photocurrent–voltage (I–V) characteristics were observed (Table 1). The maximum IPCE value increased in the order of OH17 (31%@578 nm) < OH4 (34%@473 nm) < OH1 (46%@478 nm) < OH2 (58%@484 nm) = OH7 (58%@486 nm). The Jsc value for OH2 (2.01 mA cm−2) is close to that for OH7 (1.91 mA cm−2), and is similar to that for OH1 (2.12 mA cm−2) and higher than those of OH4 (1.5 0 mA cm−2) and OH17 (1.30 mA cm−2) with two carboxyl groups. The η values increase in the order of OH17 (0.61%) < OH4 (0.67%) < OH7 (0.90%) ≤ OH1 and OH2 (1.00%). The Voc values for OH1, OH2, OH4, OH7, and OH17 are 508, 548, 470, 520, and 488 mV, respectively, depending largely on the sort of the dyes. Therefore, the photovoltaic performances of OH2 and OH7 are similar to that of OH1 and higher than those of OH4 and OH17. Here, it is worth mentioning that the Jsc and η values for OH7 with a longer alkyl chains containing a carboxyl group at the end position are slightly smaller than those for OH2.
The processes (5) and (6) affect Jsc and Voc of DSSCs, respectively.18–20 Thus, first of all, the transient absorption technique was applied to the dyes OH1, OH2, OH4, OH7, and OH17 to evaluate the rate of the charge recombination process (5). The absorption spectrum of dye cations on TiO2 needed for the transient absorption spectroscopy was determined by the spectroelectrochemical measurements with the dye-adsorbed TiO2 electrode in 0.1 M Et4NClO4/acetonitrile. A typical difference absorption spectrum for OH1 is depicted in Fig. 3, where the dye-adsorbed TiO2 electrode is biased at 0.6 V corresponding to the oxidation potential of OH1 (Eox = 0.58 V vs.Ag/AgCl) determined by cyclic voltammetry (CV). It is seen clearly that two absorption bands due to dye cation appear at around 550 and 800–1200 nm. Thus, the charge recombination kinetics was followed by monitoring the 850 nm band for the DSSC without a redox electrolyte. Fig. 4 shows the transient absorption traces for the five dyes. Significant differences in the charge recombination rate were observed for the five dyes. The recombination half-time (t50%) increases in the order of OH17 (21 μs) < OH4 (195 μs) < OH2 (1.8 ms) ≤ OH1 (2.5 ms) < OH7 (9 ms), in accord with the increasing order of the maximum IPCE values.
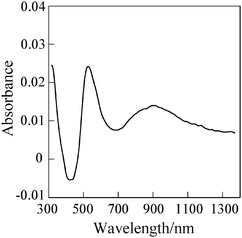 | ||
| Fig. 3 Difference absorption spectrum for OH1-adsorbed TiO2 on FTO in 0.1 M Et4NClO4/acetonitrile measured at potentials between 0 and 0.6 V, where a dye-adsorbed TiO2 on FTO as a working electrode, Pt wire as a counter electrode and Ag/AgCl as a reference electrode were used. | ||
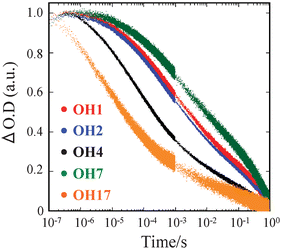 | ||
| Fig. 4 Transient absorption data monitoring charge recombination of injected electrons to TiO2 with OH1, OH2, OH4, OH7, and OH17 cations. The data were obtained by observing the decay of the 850 nm cation absorption for all five dye/TiO2 film combinations, in the absence of a redox electrolyte. | ||
The kinetics of the dark current (6) was studied by using the transient photovoltage technique. Fig. 5a shows a typical photovoltage transient for DSSC sensitized with OH1. The electron lifetimes (τe) of DSSCs based on OH1, OH2, OH4, and OH7 are plotted in Fig 5b against the Voc values obtained under different intensities of light. The τe values for OH1, OH2, OH4, or OH7 measured at around 450 mV are 11.5, 10.2, 9.1, and 15.1 ms, respectively.
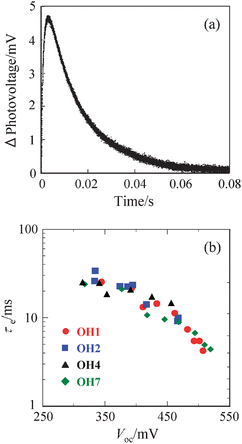 | ||
| Fig. 5 (a) Transient photovoltage decay of DSSC sensitized with OH1 and (b) electron lifetimes (τe) of DSSCs based on OH1, OH2, OH4, or OH7 under corresponding Voc values resulting from various light intensities. | ||
Discussion
On the basis of the ICT characteristics revealed by the MO calculations and the kinetics of the processes (5) and (6) estimated by the transient absorption spectroscopy and the transient photovoltage technique, the differences of photovoltaic performances among the dyes OH1, OH2, OH4, OH7, and OH17 will be discussed below from the viewpoint of configurations of the dyes adsorbed on TiO2 surface. Based on the ICT characteristics and the t50% values, possible configurations of the dye molecules adsorbed on TiO2 surface may be inferred as shown in Fig. 6. The short t50% for OH4 (195 μs) demonstrates that the cationic charge on the dialkylamino moiety in the photoexcited state is located in close proximity to the TiO2 surface because of the rigid double ester linkages with TiO2 surface for OH4, which results in the rapid recombination of electrons injected to the CB of TiO2 with dye cation. In this connection, the rigid double ester linkages with TiO2 surface prevent the cyano group of the dye from approaching the TiO2 surface, so that the intramolecular charge transfer from the dialkylamino moiety to the carboxylphenyl moiety for OH4 accompanying photoexcitation flows in the opposite direction of TiO2 surface, which leads to reduction in electron-injection yield from the excited dye to the CB of TiO2. Thus, the rapid charge recombination and the low electron injection-yield may explain smaller IPCE and Jsc values for OH4.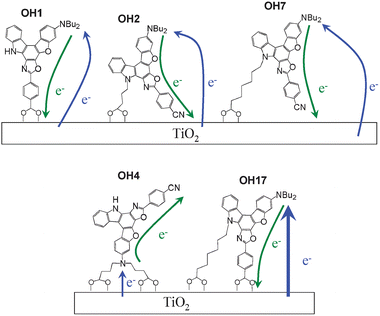 | ||
| Fig. 6 Plausible configurations of OH1, OH2, OH4, OH7, and OH17 on TiO2 surface. The green and blue arrows show electron injection from the dye molecule into TiO2 and charge recombination of injected electrons to TiO2 with dye molecule, respectively. | ||
In OH1, on the other hand, the intramolecular charge transfer takes place from the dialkylamino moiety to the carboxylphenyl moiety upon photoexcitation and the large t50% value of OH1 (2.5 ms) is comparable to those of the traditional D-π-A dye sensitizers reported earlier.19 Thus, as shown in Fig. 6, electrons in OH1 will be efficiently injected from the dye through carboxyl group to the CB of TiO2, leading to the large IPCE and Jsc values for the DSSC based on OH1.
It has been already suggested that the phenylcyano group acting as electron acceptor in OH2 and OH7 is located close to the TiO2 surface.14,15 Therefore, the dyes OH2 and OH7 can efficiently inject electrons into the TiO2 electrode through the cyano group (Fig. 6). In view of the ICT characteristics of OH2 and OH7, the increased distance between the cationic charge on the dialkylamino moiety in the photoexcited state and the TiO2 surface explains the large t50% values of OH2 (1.8 ms) and OH7 (9 ms). Thus, the high electron-injection yield and the retardation of the charge recombination for OH2 and OH7 lead to their larger Jsc values. As for the reason for the large t50% value of OH7, we presume that the longer alkyl chain containing a carboxyl group at the end position of OH7 causes the retardation of the charge recombination because of the increased separation of the cationic charge on the dialkylamino moiety of the dye from the TiO2 surface.
On the other hand, the dye OH17 with two carboxyl groups can interact strongly with TiO2 surface through double ester linkages, so that electrons will be injected from the dye to TiO2 through carboxyl group in carboxyphenyl moiety, similarly to the case of OH1. However, the Jsc or IPCE value of OH17 is smaller than that of OH1. Tian and co-workers have also reported that stronger adsorption of the dye with two carboxyl groups on the TiO2 surface could facilitate charge recombination of the injected electron and oxidized dye.21 In view of this and the smallest t50% value for OH17 (21 μs), the strong interaction with TiO2 surface for OH17 may enhance the charge recombination between the injected electron and the excited dye (dye cation), which was faster than for comparable dyes that have one carboxyl group because of close approach of the oxidized OH17 to TiO2 surface. This may be a sound reason for the low photovoltaic performance of OH17.
It was found from the transient photovoltage measurements that the τe values of DSSCs based on OH1, OH2, OH4, and OH7 are similar to one another. Ko et al. have demonstrated that the dark current (6) is effectively controllable by the modification of dye molecular structure: the introduction of hydrophobic alkyl chains and bulky substituents to dye molecular structure discourages the process (6), leading to the increased Voc.20c,d,f Judging from the close τe values for OH1, OH2, OH4, OH7, and OH17, however, the differences in Voc among the five dyes are not ascribable to the process (6). Meanwhile, it has been reported that the dipole of a dye adsorbed on TiO2 affects the electronic state of TiO2 and the Fermi level of TiO2 can be changed by adsorption of dyes having permanent dipoles.22 The directions of dipole moments of OH1, OH2, OH4, OH7, and OH17 in the adsorption state are different because of different positions of carboxyl groups on a chromophore skeleton and the number of carboxyl groups. Thus, it may be reasonable to conclude that the difference in Voc among the five dyes arises not from the process (6), but from the difference in the direction of the dipole moments of the respective dyes.
Consequently, it is revealed that the differences in photovoltaic performance among the D-π-A dye sensitizers OH1, OH2, OH4, OH7, and OH17 with carboxyl groups on different positions of a chromophore skeleton are ascribable not only to electron-injection yield from the excited dye to the CB of TiO2, but also to the recombination process between the electrons injected to the CB of TiO2 and the excited dye. The high electron-injection yield and the retardation of the charge recombination process (5) for OH2 and OH7, which are functionally-separated into an anchoring group for attachment on TiO2 surface and an electron acceptor moiety, are responsible for the high Jsc comparable to that of the D-π-A dye sensitizer OH1.
Conclusions
As a new class of donor-π-acceptor (D-π-A) dye sensitizers, a series of benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type fluorescent dyes OH1, OH2, OH4, OH7, and OH17 with carboxyl groups on different positions of a chromophore skeleton were examined as photosensitizers in DSSCs. In order to elucidate the difference of the DSSC performances among the five dyes from the viewpoint of configuration of the dyes adsorbed on TiO2 surface, kinetic studies on the reactions of electrons injected to the CB of TiO2 with dye cation and with I3− ions in solution were performed by employing the transient absorption spectroscopy and the transient photovoltage technique. Combining the results with the MO calculations explains that the high electron-injection yield and the retardation of the charge recombination for new type of D-π-A dye sensitizers OH2 and OH7 functionally-separated into an anchoring group and an electron acceptor moiety are responsible for the large Jsc values of OH2 and OH7 comparable to that of OH1 which is a traditional D-π-A dye sensitizer. We propose that, for developing high performance DSSCs, it is necessary to create a novel molecular design of organic dye sensitizers capable of controlling not only the photophysical and electrochemical properties of the dyes themselves but also the molecular orientation and arrangement of the dyes on TiO2 surface, which will be made possible by the exquisite molecular design and synthetic strategy of a new type of D-π-A dye sensitizers functionally separated into an anchoring group and an electron acceptor moiety.Experimental
Absorption spectra were taken on a Shimadzu UV-3150 spectrophotometer and fluorescence spectra were measured with a Hitachi F-4500 spectrophotometer. The fluorescence quantum yields (Φ) were determined by a Hamamatsu C9920-01 equipped with CCD by using a calibrated integrating sphere system (λex = 325 nm). Cyclic voltammograms (CVs) were recorded in MeCN/Et4NClO4 (0.1 M) solution with a three-electrode system consisting of Ag/Ag+ as a reference electrode, Pt plate as a working electrode, and Pt wire as a counter electrode, by using a Hokuto Denko HAB-151 potentiostat equipped with a functional generator. Spectroelectrochemical measurements were carried out with a Shimadzu UV-3150 spectrophotometer, where a dye-adsorbed TiO2 on FTO was used as a working electrode in 0.1 M Et4NClO4/acetonitrile with Pt wire as a counter and Ag/AgCl as a reference electrode.Computational methods
The semi-empirical calculations were carried out with the WinMOPAC Ver. 3.9 package (Fujitsu, Chiba, Japan). Geometry calculations in the ground state were made using the AM1 method.23 All geometries were completely optimized (keyword PRECISE) by the eigenvector following routine (keyword EF). Experimental absorption spectra of the five compounds were compared with their absorption data by the semi-empirical method INDO/S (intermediate neglect of differential overlap/spectroscopic).24 All INDO/S calculations were performed using a single excitation full SCF/CI (self-consistent field/configuration interaction), which includes the configuration with one electron excited from any occupied orbital to any unoccupied orbital, where 225 configurations were considered [keyword CI (15 15)].Preparation of dye-sensitized solar cells
The TiO2 electrodes used for dye-sensitized solar cells were prepared as follows. Into a powder (1.30 g) of TiO2 (P-25, Φ = 30–40 nm) in a mortar was added water (1.9 mL) in six portions with well stirring. Then, three drops of 12 M nitric acid and polyethylene glycol (80 mg) were successively added, and the mixture was well kneaded to a smooth paste. It was then applied on a fluorine-doped-tin-oxide (FTO) substrate and sintered for 30 min at 500 °C. The 7 μm thick TiO2 electrode (0.5 × 0.5 cm2 in photoactive area) was immersed into a 0.3 mM tetrahydrofuran solution of the dye for hours enough to adsorb the photosensitizer. The DSSCs were fabricated by using the dye-adsorbed TiO2 electrode thus prepared, Pt-coated glass as a counter electrode, and a solution of 0.05 M iodine, 0.1 M lithium iodide, and 1,2-dimethyl-3-n-propylimidazolium iodide in acetonitrile as electrolyte. The photocurrent–voltage characteristics were measured using a potentiostat under a simulated solar light (AM 1.5, 61 mW cm−2). IPCE spectra were measured under monochromatic irradiation with a tungsten–halogen lamp and a monochromator. The dye-coated film was immersed in a mixture of THF solvent, DMSF and 1 M NaOH aq (5∶4∶1), which was used to determine the amount of dye molecule adsorbed onto the film by measuring the absorbance. The quantification of each dye was made based on the λmax and the molar extinction coefficient of each dye in the above solution. Absorption spectra of the dye-adsorbed TiO2 films adsorbed by dyes were measured in the diffuse-reflection mode by a Shimadzu UV-3150 spectrophotometer with a calibrated integrating sphere system ISR-3100.
Transient absorption spectroscopy
The transient absorption measurements were performed by the conventional pump and probe technique. The dyes adsorbed on TiO2 film were excited at 480 nm for all five dyes with pulses from a nitrogen laser pumped dye laser (1 ns pulse duration, 6 Hz repetition, ∼44 μJ cm−2 at 480 nm). The resulting photoinduced change in optical density was monitored by employing the probe light of 850 nm from a light emitting diode corresponding to dye cation absorption determined by in situ spectroelectrochemical measurement of dye adsorbed on TiO2 film, a Hamamatsu Photonics C8366 PIN photodiode as a photosensor, and an IWATSU DS-4372 oscilloscope.Transient photovoltage technique
Transient photovoltage measurements in the DSSCs employed a 100 μs exciting pulse generated by a ring of light emitting diode (λ = 475 nm), which is controlled by a pulse motor. The pulse was incident on the TiO2 side of the device and its intensity was controlled to keep the modulation of the voltage below 5 mV. White bias light impinging the cell from the same direction was also supplied by halogen lamp (150 W). The transient photovoltage decay was obtained from an oscilloscope (IWATSU DS-4372) with an RC circuit (τ = 2.2 s) through an NF 5307 voltage amplifier.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) (19350094) and for Young Scientist (B) (22750179) from the Japan Society for the Promotion of Science (JSPS), by General Sekiyu Research & Development Encouragement & Assistance Foundation, and by the Kurata Memorial Hitachi Science and Technology Foundation.References
- (a) N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338 CrossRef CAS; (b) N. Robertson, Angew. Chem., Int. Ed., 2008, 47, 1012 CrossRef CAS.
- Z. Chen, F. Li and C. Huang, Curr. Org. Chem., 2007, 11, 1241 CrossRef CAS.
- P. Xie and F. Guo, Curr. Org. Chem., 2007, 11, 1272 CrossRef CAS.
- Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903 CrossRef CAS.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC.
- (a) Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2008, 112, 17011 CrossRef CAS; (b) D. P. Hagberg, J.-H. Yum, H. Lee, F. D. Angelis, T. Marinado, K. M. Karlsson, R. Humphry-Baker, L. Sun, A. Hagfeldt, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2008, 130, 6259 CrossRef CAS; (c) K. R. J. Thomas, J. T. Lin, Y.-C. Hsua and K.-C. Ho, Chem. Commun., 2005, 4098 RSC; (d) G. Zhou, N. Pschirer, J. C. Schöneboom, F. Eickemeyer, M. Baumgarten and K. Müllen, Chem. Mater., 2008, 20, 1808 CrossRef CAS; (e) G. Li, K.-J. Jiang, Y.-F. Li, S.-L. Li and L.-M. Yang, J. Phys. Chem. C, 2008, 112, 11591 CrossRef CAS.
- (a) N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256 CrossRef CAS; (b) S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Frantacci, F. D. Angelis, D. D. Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS; (c) H. Choi, C. Baik, S. O. Kang, J. Ko, M.-S. Kang, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 327 CrossRef CAS; (d) H. Qin, S. Wenger, M. Xu, F. Gao, X. Jing, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 9202 CrossRef CAS.
- (a) T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218 CrossRef CAS; (b) T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137 CrossRef CAS; (c) Y. Shibano, T. Umeyama, Y. Matano and H. Imahori, Org. Lett., 2007, 9, 1971 CrossRef CAS; (d) T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137 CrossRef CAS; (e) W. H. Howie, F. Claeyssens, H. Miura and L. M. Peter, J. Am. Chem. Soc., 2008, 130, 1367 CrossRef CAS; (f) S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Péchy and M. Grätzel, Chem. Commun., 2008, 5194 RSC.
- (a) Y. Chen, Z. Zeng, C. Li, W. Wang, X. Wang and B. Zhang, New J. Chem., 2005, 29, 773 RSC; (b) S. Tatay, S. A. Haque, B. O'Regan, J. R. Durrant, W. J. H. Verhees, J. M. Kroon, A. Vidal-Ferran, P. Gaviña and E. Palomares, J. Mater. Chem., 2007, 17, 3037 RSC; (c) J.-H. Yum, P. Walter, S. Huber, S. Rentsch, T. Geiger, F. Nüesch, F. D. Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320 CrossRef CAS; (d) A. Burke, L. Schmidt-Mende, S. Ito and M. Grätzel, Chem. Commun., 2007, 234 RSC; (e) J. Rochford, D. Chu, A. Hagfeldt and E. Galoppini, J. Am. Chem. Soc., 2007, 129, 4655 CrossRef CAS; (f) S. Eu, S. Hayashi, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 4396 CrossRef CAS; (g) S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846 CrossRef CAS; (h) A. Burke, S. Ito, H. Snaith, U. Bach, J. Kwiatkowski and M. Grätzel, Nano Lett., 2008, 8, 977 CrossRef CAS.
- (a) E. Palomares, M. V. Martinez-Diaz, S. A. Haque, T. Torres and J. R. Durrant, Chem. Commun., 2004, 2112 RSC; (b) A. Moranderia, I. López-Duarte, M. V. Martinez-Diaz, B. O'Regan, C. Shuttle, N. A. Haji-Zainulabidin, T. Torres, E. Palomares and J. R. Durrant, J. Am. Chem. Soc., 2007, 129, 9250 CrossRef CAS; (c) P. Y. Reddy, L. Giribabu, C. Lyness, H. J. Snaith, C. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J.-H. Yum, K. Kalyanasundaram, M. Grätzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373 CrossRef CAS; (d) J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martinez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS; (e) B. C. O'Regan, I. López-Duarte, M. V. Martinez-Diaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 2906 CrossRef CAS; (f) S. Eu, T. Katoh, T. Umeyama, Y. Matano and H. Imahori, Dalton Trans., 2008, 5476 RSC.
- (a) B. Liu, W. Zhu, Q. Zhang, W. Wu, M. Xu, Z. Ning, Y. Xie and H. Tian, Chem. Commun., 2009, 1766 RSC; (b) G. Li, Y.-F. Zhou, X.-B. Cao, P. Bao, K.-J. Jiang, Y. Lin and L.-M. Yang, Chem. Commun., 2009, 2201 RSC; (c) H. Tian, X. Yang, J. Cong, R. Chen, J. Liu, Y. Hao, A. Hagfeldt and L. Sun, Chem. Commun., 2009, 6288 RSC; (d) H. Im, S. Kim, C. Park, S.-H. Jang, C.-J. Kim, K. Kim, N.-G. Park and C. Kim, Chem. Commun., 2010, 46, 1335 RSC; (e) W. Wu, J. Yang, J. Hua, J. Tang, L. Zhang, Y. Long and H. Tian, J. Mater. Chem., 2010, 20, 1772 RSC; (f) H. Choi, J.-J. Kim, K. Song, J. Ko, Md. K. Nazeeruddin and M. Grätzel, J. Mater. Chem., 2010, 20, 3280 RSC; (g) C. Teng, X. Yang, C. Yang, H. Tian, S. Li, X. Wang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 11305 CrossRef CAS; (h) H. Choi, I. Raabe, D. Kim, F. Teocoli, C. Kim, K. Song, J.-H. Yum, J. Ko, Md. K. Nazeeruddin and M. Grätzel, Chem.–Eur. J., 2010, 16, 1193 CrossRef CAS; (i) C.-H. Chen, Y.-C. Hsu, H.-H. Chou, K. R. J. Thomas, J. T. Lin and C.-P. Hsu, Chem.–Eur. J., 2010, 16, 3184 CrossRef CAS.
- (a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS; (b) A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef CAS; (c) U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583 CrossRef; (d) M. Grätzel, Nature, 2001, 414, 338 CrossRef CAS.
- (a) Y. Ooyama, A. Ishii, Y. Kagawa, I. Imae and Y. Harima, New J. Chem., 2007, 31, 2076 RSC; (b) Y. Ooyama, Y. Shimada, Y. Kagawa, I. Imae and Y. Harima, Org. Biomol. Chem., 2007, 5, 2046 RSC.
- (a) Y. Ooyama, Y. Shimada, Y. Kagawa, Y. Yamada, I. Imae, K. Komaguchi and Y. Harima, Tetrahedron Lett., 2007, 48, 9167 CrossRef; (b) Y. Ooyama, Y. Shimada, A. Ishii, G. Ito, Yu. Kagawa, I. Imae, K. Komaguchi and Y. Harima, J. Photochem. Photobiol., A, 2009, 203, 177 CrossRef CAS.
- Y. Ooyama, S. Inoue, R. Asada, G. Ito, K. Kushimoto, K. Komaguchi, I. Imae and Y. Harima, Eur. J. Org. Chem., 2010, 92 CAS.
- Y. Hao, X. Yang, J. Cong, H. Tian, A. Hagfeldt and L. Sun, Chem. Commun., 2009, 4031 RSC.
- (a) S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538 CrossRef CAS; (b) J. N. Clifford, E. Palomares, Md. K. Nazeeruddin, M. Grätzel, J. Nelson, X. Li, N. J. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225 CrossRef CAS; (c) N. Hirata, J.-J. Lagref, E. J. Palomares, J. R. Durrant, Md. K. Nazeeruddin, M. Grätzel and D. D. Censo, Chem.–Eur. J., 2004, 10, 595 CrossRef CAS; (d) J. E. Kroeze, N. Hirata, S. Koops, Md. K. Nazeeruddin, L. Schmidt-Mende, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2006, 128, 16376 CrossRef CAS; (e) Z.-S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Frube and K. Hara, Chem. Mater., 2008, 20, 3993 CrossRef CAS.
- (a) S. A. Haque, S. Handa, K. Peter, E. Palomares, M. Thelakkat and J. R. Durrant, Angew. Chem., Int. Ed., 2005, 44, 5740 CrossRef CAS; (b) S. Tan, J. Zhai, H. Fang, T. Jiu, J. Ge, Y. Li, L. Jiang and D. Zhu, Chem.–Eur. J., 2005, 11, 6372 CrossRef CAS; (c) S. Handa, H. Wietasch, M. Thelakkat, J. R. Durrant and S. A. Haque, Chem. Commun., 2007, 1725 RSC; (d) A. Forneli, M. Olanells, M. A. Sarmentero, E. Matyinez-Ferrero, B. C. O'Regan, P. Ballester and E. Palomares, J. Mater. Chem., 2008, 18, 1652 RSC.
- (a) S. Nakade, T. Kanzaki, Y. Wada and S. Yanagida, Langmuir, 2005, 21, 10803 CrossRef CAS; (b) G. Boschloo, L. Häggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144 CrossRef CAS; (c) S. Kim, D. Kim, H. Choi, M.-S. Kang, K. Song, S. O. kang and J. Ko, Chem. Commun., 2008, 4951 RSC; (d) C. Lee, J.-H. Yum, H. Choi, S. O. Kang, J. Ko, R. Humphry-Baker, M. Grätzel and Md. K. Nazeeruddin, Inorg. Chem., 2008, 47, 2267 CrossRef CAS; (e) D. Kuang, S. Uchida, R. Humphry-Baker, S. K. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923 CrossRef CAS; (f) D. Kim, M.-S. Kang, K. Song, S. O. Kang and J. Ko, Tetrahedron, 2008, 64, 10417 CrossRef CAS; (g) S. Wenger, P.-A. Bouit, Q. Chen, J. Teuscher, D. D. Censo, R. Humphry-Baker, J.-E. Moser, J. L. Delgado, N. Martin, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 5164 CrossRef CAS.
- Q.-H. Yao, F.-H. Meng, F.-Y. Li, H. Tian and C.-H. Huang, J. Mater. Chem., 2003, 13, 1048 RSC.
- S. Rühle, M. Greenshtein, S.-G. Chen, A. Merson, H. Pizem, C. S. Sukenik, D. Cahen and A. Zaban, J. Phys. Chem. B, 2005, 109, 18907 CrossRef.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef.
- (a) J. E. Ridley and M. C. Zerner, Theor. Chim. Acta, 1973, 32, 111 CrossRef CAS; (b) J. E. Ridley and M. C. Zerner, Theor. Chim. Acta, 1976, 42, 223 CrossRef CAS; (c) A. D. Bacon and M. C. Zerner, Theor. Chim. Acta, 1979, 53, 21 CrossRef CAS; (d) H. A. Kurtz, J. J. P. Stewart and D. M. Dieter, J. Comput. Chem., 1990, 11, 82 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |