Correlation between gel-forming ability, supramolecular aggregates and main-chain conformation of dendronized polymer gelators†
Zijian
Zhang
a,
Miao
Yang
a,
Changyu
Hu
a,
Bo
Liu
a,
Minbiao
Hu
a,
Xinjun
Zhang
a,
Wei
Wang
*a,
Yunchun
Zhou
b and
Hanfu
Wang
b
aCenter for Synthetic Soft Materials, The Key Laboratory of Functional Polymer Materials of Ministry of Education and Institute of Polymer Chemistry, Nankai University, Tianjin 300071, China. E-mail: weiwang@nankai.edu.cn; dishman@mail.nankai.edu.cn; miaoyang@rohmhaas.com; zhangxj@mail.nankai.edu.cn; huchangyu168@mail.nankai.edu.cn; liubo-123@mail.nankai.edu.cn; hmb@mail.nankai.edu.cn; weiwang@nankai.edu.cn
bState Key Laboratory of Polymer Physics and Chemistry and Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun 130022, China. E-mail: hfwang@ciac.jl.cn; yczhou@ciac.jl.cn
First published on 11th October 2010
Abstract
Dendronized polymer (DP) gelators were prepared via complexation of a dendritic gelator of the poly(urethane amide) dendron, which contains an ionizing carboxyl group on the apexes, with three positively charged polyelectrolytes. All these complexes could form organogels in toluene, and their gel properties were studied through determination of their minimum gelation concentration and characterization of the networked supramolecular structures formed in their gel-phase using TEM, AFM and SAXS. Our findings showed great differences in the gel-forming ability and supramolecular structure among these gelators. A simple simulation on the conformations of the three polyelectrolytes illustrated that the differences came mainly from the polyelectrolyte conformations which produced different distribution and orientation of the dendritic gelators along the backbones. When the backbone could induce the dendritic gelators to form a pre-ordered structure before gelation, the DP gelator would form nice supramolecular aggregates showing an enhanced gel-forming ability. Without this, the gel-forming ability would not be enhanced.
Introduction
As an important part of soft materials,1 gels, which possess both the appearance of liquid and the property of solid,2 have been applied in various fields owing to their unique properties. The types of gels are divided into polymer gels3–5 and low-mass-molecule (LMM) gels6–8 in accordance with gelator molecular weight. Polymer gels, which are constructed either by covalent cross-linking to get chemical gels, or through physical changes contributed by non-covalent interactions between macromolecules to form physical gels, have been extensively investigated and widely applied.3–5LMM gels, which are always supramolecular,6–8 are usually initiated by the self-assembly of LMM gelators through non-covalent interactions, such as hydrogen bonds,9 π–π stacking,10 donor-accepter interactions,11etc., to construct physical gels. It has been reported that some supramolecular polymers, which were constructed by LMM motifs through non-covalent interactions, have formed gels as a result of a phase separation and crystallization process.12 The most interesting functionality is that they could be destroyed and then rebuilt up under special conditions. The previous studies have demonstrated that the formation process of LMM gels can be divided into three major steps as shown in Fig. 1.7b,c Firstly the gelator molecules form linear supramolecular polymers initiated by intermolecular forces, and then they aggregate together into fibers or other supramolecular aggregates which are applied at the third step to construct a gel network, which could trap and then immobilize solvent molecules inside. In recent years, LMM gels have gained attention for their unique assembly properties in supramolecular chemistry and potential applications in various fields, especially in biological science owing to their degradable ability.13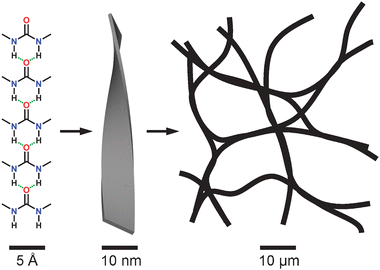 | ||
| Fig. 1 Schematic representation for the primary, secondary, and tertiary steps of the gelation process of a self-assembled physical gel.7c | ||
Due to well-defined architectures and diverse functionalities, dendrimers and dendrons have been widely applied in the study of supramolecular assembly.14 It has been reported that some dendrons, with various decorations on their apexes or peripheries, could form gel-phase materials mainly in organic media.15 They can be called dendritic gelators with a wedge shape. Most of them contain amide and/or urethane groups which provide multiple intermolecular hydrogen bond sites and peripheral alkyl chains which provide interactions with organic solvents. During gelation, the formation of multiple hydrogen bonds is the advantage and key factor for dendrons in constructing supramolecular aggregates, such as ribbons,15b which could braid into networks and finally create a gel. In other words, these dendritic gelators form a gel-phase normally following the steps shown in Fig. 1. To promote the gel-formation ability of such dendritic gelators, it has been reported that the dumbbell-shaped dimers16 and diblock dendrimers17 formed by covalent or non-covalent connections of these dendrons with bifunctionalized motifs can enhance the gel-forming ability of dendritic gelators. Meanwhile, dendron-polymer gelators, which were prepared by linking a dendron to a linear polymer,18 have also been studied. In one of our previous works we created the codendrimer gelators by covalently linking poly(urethane amide) (PUA) dendrons and poly(methallyl dichloride) (PMDC) dendrons together, and found that their gel-forming ability was expedited with increasing generation of both the dendrons because higher polarity and multiple hydrogen bonds can promote the aggregation ability of the gelator.17a It has been reported that some dendronized polymers (DPs) which were created by covalently attaching dendrons to linear polymer backbones show attractive gel-forming ability.19DP can be also prepared through complexation of dendritic molecules with polyelectrolytes.20 In some of our other previous research,21 we have constructed a DP gelator applying this kind of complexation method to attach second generation (g2) PUA dendrons to the backbone of a polyelectrolyte [poly(diallyldimethylammonium chloride), abbreviated as PDADMA]. Importantly, we found that the DP gelator (abbreviated as g2-PUA-PDADMA) displayed a gel-forming ability stronger than the parent g2-PUA gelator because the main chain of PDADMA assisted g2-PUA dendrons to form a pre-ordered one-dimensional structure before gelation. In this work we will report our further study concerning the effect of main-chain conformation of the polyelectrolytes in the gelators of the g2-PUA dendron-polyelectrolyte complexes on their gel-forming ability. Three polyelectrolytes: PDADMA, poly(allylamine hydrochloride) (abbreviated as PAH) and quaternized poly(4-vinylpyridine) (abbreviated as QP4VP) will be used to construct three DP gelators of g2-PUA-polyelectrolyte complexes (see Fig. 2) mainly due to different main-chain structures as well as conformations. These gelators are abbreviated as g2-PUA-PDADMA, g2-PUA-PAH and g2-PUA-QP4VP, respectively. In our work, their gel-forming ability will be evaluated via determination of their minimum gelation concentration (cmgc) and their supramolecular aggregates in their gel-phase in toluene will be characterized using transmission electron microscopy (TEM), atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS). The difference in their gel-forming ability and supramolecular aggregates can be reasonably explained in consideration of the effect of the main-chain conformation of these polyelectrolytes on the formation of pre-ordered one-dimensional structure of the dendritic gelators. The results reported in this work can help us understand the macromolecular effect in the gelation processes of such the DP gelators.
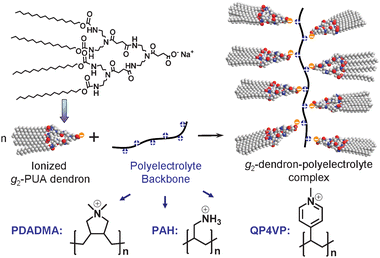 | ||
| Fig. 2 Schematic synthetic approach of g2-PUA-polyelectrolyte complexes formed via complexation of g2-PUA dendron with PDADMA, PAH and QP4VP polyelectrolytes, respectively, via ionic interactions. | ||
Results and discussion
Gel-forming ability
The method used for the synthesis of three kinds of dendron-polyelectrolyte complexes is the same as that used in a previous study21 and the characterization is summarized in the ESI†. Fig. 2 exhibits the synthesis route and the chemical structures of the three polyelectrolytes. Three DP gelators (g2-PUA-PDADMA, g2-PUA-PAH and g2-PUA-QP4VP) and g2-PUA dendron gelator are capable of forming gel in toluene.Fig. 3 shows the immobilized feature of the gelled solutions characterized by the inverting-bottle method. The gels of the g2-PUA, g2-PUA-PDADMA and g2-PUA-PAH gelators are colorless and transparent, but the gel of the g2-PUA-QP4VP gelator is slightly opaque and yellowish. Their cmgc were determined after the solutions were gelled for one or two days.22 We found cmgc = 0.5 g L−1 for the g2-PUA-PDADMA gelator, cmgc = 2.0 g L−1 for the g2-PUA-PAH gelator, cmgc = 9.0 g L−1 for the g2-PUA-QP4VP gelator after their solutions were gelled for one day, while cmgc = 8.0 g L−1 for the g2-PUA gelator which was detected after a gelation time of two days. Clearly, the g2-PUA-PDADMA gelator exhibits the lowest cmgc, while the g2-PUA-QP4VP gelator displays the highest cmgc, which is slightly higher than cmgc of the g2-PUA gelator. Now we can conclude that the gel-forming ability of the g2-PUA gelator is enhanced via the complexation with PDADMA and PAH but is somehow worsened with QP4VP.
 | ||
| Fig. 3 Photographs of the gels in toluene with a concentration near their cmgc. (A) g2-PUA gelator on 8.0 g L−1 after being gelled for two days; (B) g2-PUA-PDADMA gelator on 0.5 g L−1 after being gelled for one day; (C) g2-PUA-PAH gelator on 2.0 g L−1 after being gelled for one day; (D) g2-PUA-QP4VP gelator on 9.0 g L−1 after being gelled for one day. | ||
Networked supramolecular aggregates
The supramolecular morphology formed in these gel-phases was further characterized by TEM and AFM. The TEM images in Fig. 4 depict complex networks constructed by fiber- or ribbon-like aggregates of the four gelators. The fine aggregates are the common feature of these gels and are also the origin of the formation of the transparent gels. Nevertheless, we can still see some differences between the aggregates of these gels. The gels of the g2-PUA and g2-PUA-PDADMA gelators show a sparse network formed by braiding and unbraiding the long and wide ribbons (see Fig. 4A and B).21 In the g2-PUA gel-phase there are some plate-like aggregates around the ribbons. They possibly correspond to the incomplete assemblies of the g2-PUA molecules, while the plate-like aggregates cannot be found in the gels of other gelators. This phenomenon is associated with a fact that during gelation the self-assembly ability of the DP gelators is higher than that of the parent dendron gelator alone. This is a key point which we will focus on in this work. The TEM image of the g2-PUA-PAH gel shows a dense network with short and fine fiber-like aggregates, as shown in Fig. 4C. It is hard to identify how the aggregates construct the network. Fig. 4D shows a network of the aggregates formed in the gel of the g2-PUA-QP4VP gelator. Clearly, the higgledy-piggledy network is constructed by the irregular aggregates with different lengths and widths. In this TEM image we can see some coarse aggregates (the dark dots in the image) with a size in a few hundred nanometres. This is the reason why the g2-PUA-QP4VP gel is slightly opaque.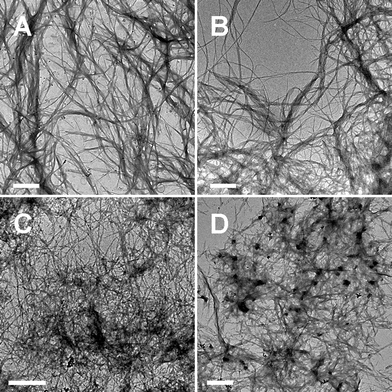 | ||
| Fig. 4 TEM images showing a network structure of supramolecular fibers or ribbons formed in the gel-phase of (A) g2-PUA, (B) g2-PUA-PDADMA, (C) g2-PUA-PAH and (D) g2-PUA-QP4VP. The scale bar is 1 μm. | ||
We also have carried out AFM characterizations to see details of the aggregates formed in the gel-phases of the three DP gelators, as shown in Fig. 5. Fig. S6 to S8 in the ESI† display the corresponding three-dimensional feature of the aggregate morphologies. Fig. 5A shows straight ribbon-like aggregates formed in the g2-PUA-PDADMA gel. They can twist and braid to build up a gel network. We could clearly see the edges for these ribbons and discover the layer-by-layer stacking of thinner ribbons (also see Fig. S6 in ESI†), which could be the reason for the formation of entangled and thicker ribbons. From the surface profile of the fine ribbon shown in Fig. 6A, we could see that the stacking ribbon has a width of ca. 110 nm and a thickness of ca. 30 nm. Fig. 5B displays the fine and thick aggregates formed in the g2-PUA-PAH gel. They are curved, meaning they should be softer than the aggregates formed in the g2-PUA-PDADMA gel. From the images we could see that the thick aggregates are composed of several fine aggregates. In other words, these fine aggregates braid into the thick aggregates. The fine aggregates also show a feature of ribbon-like structure, as shown in Fig. 6B, with a width of ca. 43 nm and a height of ca. 8 nm. For the g2-PUA-QP4VP gel, the features of the aggregates are similar to those we found in the g2-PUA-PAH gels (see Fig. 5C). Relatively speaking, the fine and thick aggregates are slightly stiffer than those formed in the g2-PUA-PAH gel. These fine aggregates also have a ribbon-like structure, as shown by the surface profile of a fine aggregate in Fig. 6C. This ribbon aggregate has a ca. 44 nm width and a ca. 12 nm thickness. In the marked area in Fig. 5C we can see some irregular areas. This means that the elongational growth of the aggregates seemingly lost their way. In summary, these TEM and AFM observations undoubtedly illustrate a difference in the ability of these gelators assembling into ordered supramolecular aggregates which finally generate the network to immobilize solvent molecules. This is evidence to demonstrate the reason why these gelators have different gel-forming abilities at a supramolecular level.
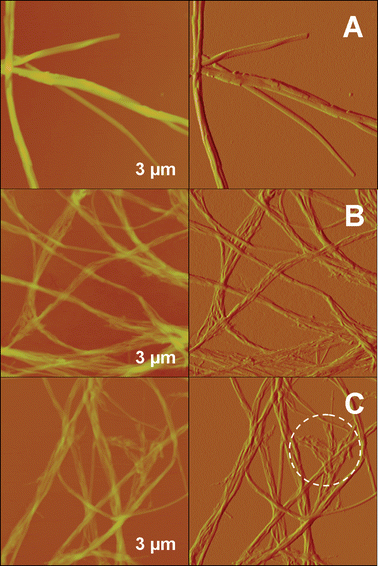 | ||
| Fig. 5 AFM height (on the right side) and amplitude (on the left side) images on 3 × 3 μm2 of dried-gel samples of (A) g2-PUA-PDADMA, (B) g2-PUA-PAH and (C) g2-PUA-QP4VP. | ||
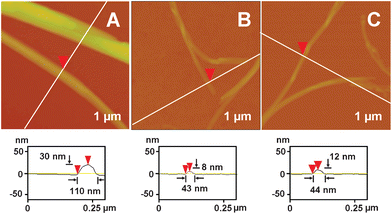 | ||
| Fig. 6 AFM height images on 1 × 1 μm2 and surface profiles of fine ribbons formed in gels of (A) g2-PUA-PDADMA, (B) g2-PUA-PAH and (C) g2-PUA-QP4VP. | ||
Ordered supramolecular structures in aggregates
To have a better understanding on the supramolecular aggregates, we have carried out SAXS experiments on dried gel samples of the gels of the three DP gelators. Fig. 7 shows the Iq2 ∼ q plots obtained in our SAXS experiments. (I = scattering intensity and q = scattering vector, where q = 4π sin θ/λ, in which 2θ is the scattering angle and λ = 0.154 nm is the wavelength of the radiation.) The SAXS data provides evidence to show ordered structures24 which are schematically illustrated by the suggested stacking models of supramolecular arrangements of the DP gelators in the aggregates. In the Iq2 ∼ q plot of Fig. 7A we can clearly see two scattering peaks, pointed out by the arrows 1 and 2, at q1 = 0.84 nm−1 and q2 = 1.68 nm−1, respectively, for the g2-PUA-PDADMA sample. The ratio q1/q2 ≈ 1/2 and the first peak position at q1 = 0.84 nm−1 suggest a layer structure with a long period of L = 7.37 nm. Meanwhile, we can also see a middle-strong scattering signal, as pointed out by arrow 1′, at q3 = 3.97 nm−1. Its d-spacing is L′ = 1.58 nm corresponding to the width of the g2 dendron. In the Iq2 ∼ q plot of Fig. 7B we can also see two scattering peaks, pointed out by the arrows 1 and 1′, at q1 = 0.99 nm−1 and q1 = 3.70 nm−1 for the g2-PUA-PAH sample. The corresponding d-spacings are L = 6.35 nm and L′ = 1.70 nm, respectively. For the g2-PUA-QP4VP gels, we can only find a wider scattering peak at q1 = 1.01 nm−1 which corresponds to a d-spacing L = 6.28 nm, but no other scattering peaks at larger q region, as shown in the Iq2 ∼ q plot of Fig. 7C.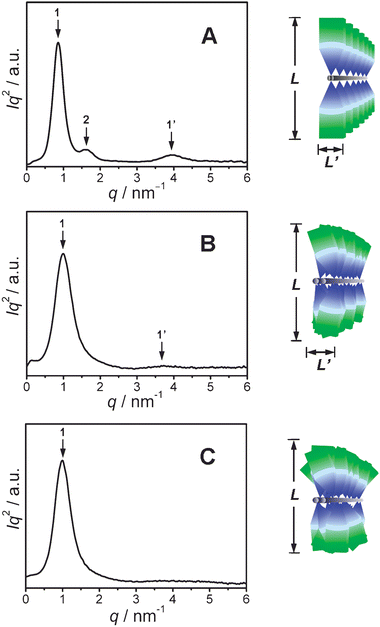 | ||
| Fig. 7 SAXS data obtained for dried gels of the DP gelators, g2-PUA-PDADMA (A), g2-PUA-PAH (B) and g2-PUA-QP4VP (C) and suggested models showing different supramolecular arrangements of g2-PUA dendron along the polymer backbones. | ||
These results indicate that the dendron molecules stack together along the polymer backbone to form ordered a supramolecular structure in aggregates. The ordered structure has a dimension L corresponding to a thickness of a layered structure within aggregates and another dimension L′ corresponding to a width of individual supramolecules within the layers. The suggested models of the ordered structure shows the dendron arrangement along macromolecule backbones in the aggregates, as schematically shown on the right of the figure. The drawing on the right of Fig. 7A suggests a highly ordered stacking structure of the g2-PUA-PDADMA dendronized polymer. In this case the polymer main chains can help dendritic gelators arrange together in a face-to-face stacking manner along the polymer backbone to form highly ordered structure within aggregates as well as within layers. So in the SAXS experiment we can detect a relatively narrow first-order scattering peak plus a second-order peak at low q region and another narrow peak at large q region. The stacking structure of the g2-PUA dendrons along PAH backbone will be not as ordered as that of the g2-PUA dendron along the PDADMA backbone, as shown schematically on the right of Fig. 7B. This is the reason that we detect only two wider peaks which correspond to the dimensions, L and L′ of the dendron-stacking structure. The case of the g2-PUA-QP4VP gelator becomes worse. The face-to-face stacking between dendritic gelators is almost interrupted, so the g2-PUA-QP4VP gelator can form the less ordered layers in the aggregates but disorder supramolecular arrangement in the layers. This is the reason that we detect only one wider peak in lower q region. From the SAXS results and relevant discussion we can see the face-to-face stacking of the dendron gelators along the polymer backbone plays an important role in the formation of highly ordered structures within aggregates as well as within layers.
Main-chain conformation and pre-ordered structure
Now, it is important to understand why these gelators have the different gel-forming abilities at a molecular level. In our previous work we have studied the gel-forming ability of the g2-PUA and g2-PUA-PDADMA gelators. We have evidently illustrated that the ability of the g2-PUA molecules assembling into ribbon-like aggregates via the formation of multiple hydrogen bonds between molecules is the origin of gelation of the g2-PUA-PDADMA gelator. Once the g2-PUA molecules are attached on the PDADMA backbone their gel-forming ability can be greatly enhanced because they can form a pre-ordered structure along the backbone. It is the pre-ordered arrangement that promotes the attached g2-PUA molecules easily and fast assembling into the supramolecular aggregate. It is natural to consider the influence of the main-chain conformation of the polyelectrolytes on the formation of the pre-ordered structure. So, we performed a simple simulation on the conformations of three kinds of polyelectrolytes to have a deeper understanding on the distinction of their gel-forming ability.Fig. 8 shows the simulated results obtained from 30 repeating units of the three polyelectrolytes, whose energy is minimized under the MMFF94 force field. In this simulation chain conformation of the three polyelectrolytes is stretched because of strong electrostatic interaction of the charges along the backbone. Meanwhile, we assume that the volume effect of counter ions is negligible.23 This assumption may be acceptable because we only want to compare the chain conformation of the three polyelectrolytes in the same condition. This is also because the strength of ionic interactions between dendritic gelators and polyelectrolytes is extremely strong in organic solvents,25 so that the influence on ionic interactions in different DP complexes can be ignored. Fig. 8A presents the side-view and top-view of the chain conformation of the three polyelectrolytes and Fig. 8B depicts the possible conformations of the three dendron-polyelectrolyte complexes in solution. The common feature of the polyelectrolyte conformation is that it is intrinsically stiff, mainly due to repulsive force between charges. The blue dots refer to the nitrogen in the cationic residues. Their distribution and orientation along the backbone is the major concern in this study. PDADMA forms a flat conformation and the cationic residues orderly distribute on the two edges of the plate and the distance between adjacent cations is about 1.0 nm on average. Such the conformation is consistent with the result reported in the previous studies.23 Because the intrinsic feature of such main-chain conformations, PAH and QP4VP should generate cylindrical backbones along which the cationic residues oriented disorderly. Due to the differences in chain stiffness and side group size, the diameter of the QP4VP cylinder is about 1.2 nm, larger than that of PAH (about 0.5 nm). With these simulation results we can reconstruct the rational chain conformations of the three dendron-polyelectrolyte complexes in solution, as shown in Fig. 8B. It is very important to emphasize that the dendrons along the PDADMA may relatively easily produce a pre-ordered structure, while it is hard for the dendrons attached on the PAH and QP4VP backbones. Such the chain conformations should be able to be applied to explain the differences in the gel-forming ability as well as supramolecular structure or morphology of the three DP gelators.
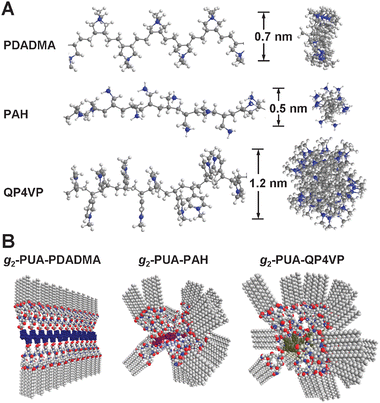 | ||
| Fig. 8 (A) Side-view and top-view of the main-chain conformations of PDADMA, PAH and QP4VP polyelectrolytes obtained via a simple simulation based on MMFF94 force field with 30 units for each polyelectrolyte. The blue dots refer to the nitrogen cations. (B) Suggested conformations of the single complexes in solution. | ||
Discussion on formation process of gel-phase
Fig. 9 represents the suggested steps of a single DP gelator joining a ribbon-like aggregate which have been experimentally found. In our previous studies,17a,21 we have indicated that the formation of the ribbon-like aggregates can maximize the multiple hydrogen bonding between adjacent PUA dendrons,26 as shown in Fig. 9, because they are the force driving the dendron into assembling an aggregate formation. It is reasonable, therefore, to suggest that the DP gelators with a random distribution of the dendrons along the backbone should at first adjust the dendron orientation to form a pre-ordered conformation (i.e. Step A), and then join the ribbons (i.e. Step B). The multiple hydrogen-bonds drive the elongational growth of ribbon-like aggregations, and the lateral growth in aggregates to form a ribbon is driven by the van der Waals interactions between alkyl chains of the dendrons, in which the stacking tacticity of dendrons plays the key factor. It is clear that during Step A there is a competition between the reduction in polyelectrolyte conformation entropy and the increase in enthalpy caused by the formation of hydrogen bonds.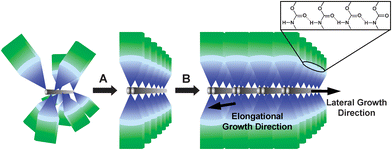 | ||
| Fig. 9 Schematic representation of the suggested steps of a DP gelator assembling into a ribbon-like aggregate during gelation. Step A is a transformation of random-to-pre-ordered distribution and orientation of dendrons along the polyelectrolyte backbone, and then two chains wrap together to form pre-ordered complexes. Step B is a process of the pre-ordered complexes joining into the ribbon resulting in elongational and lateral growths. The hydrogen bonds between adjacent dendrons along backbones are presented. | ||
Due to the special chain conformation of PDADMA, two chains of the g2-PUA-PDADMA gelator can wrap together, so that the distance between g2-PUA dendrons were suitable for hydrogen-bond formation, about 0.47 nm.24 Then the g2-PUA dendrons in the g2-PUA-PDADMA gelator could form a pre-ordered structure quickly and orderly for elongational and lateral growths to form long and wide ribbons (also see Fig. 6B). Accordingly, the individual g2-PUA-PDADMA gelator arrange into an ordered supramolecular structure in the ribbons. Therefore, the g2-PUA-PDADMA gelator demonstrates a relatively strong gel-forming ability.
For the g2-PUA-PAH and g2-PUA-QP4VP gelators, we consider that the conformations of PAH and QP4VP are not so ordered as PDADMA, so the step A should be necessary but more intricate. In this step, the main-chain adjusts its conformation and several complex molecules intertwine together to let the g2-PUA dendrons to arrange orderly. Finally, the pre-ordered complexes join the ribbon-like aggregates in which hydrogen bonding is maximized. Clearly, PAH backbone is more flexible than QP4VP owing to owning the small side group. This difference creates a dissimilar ability for forming ribbon-like aggregates. As a result, the g2-PUA-PAH gelator can overcome the first step to form a pre-ordered structure of multiple molecules relatively easier than the g2-PUA-QP4VP gelator, through tuning the conformation or stacking several complexes together. Due to these factors, the elongational and lateral growths of the ribbon-like aggregates of the g2-PUA-PAH and g2-PUA-QP4VP gelators can be greatly restricted, giving rise to a decrease of the ribbon length and width in comparison with the g2-PUA-PDADMA gelator. Therefore, the g2-PUA-PAH gelator presents a cmgc which is a little higher than the g2-PUA-PDADMA gelator but lower than the g2-PUA-QP4VP gelator. Now, we can understand from the molecular level why the gel-forming ability of the g2-PUA-PDADMA gelator is strongest and the g2-PUA-QP4VP gelator is worst.
Conclusions
In summary, we have prepared three DP gelators through complexation of the g2-PUA gelator molecules with three polyelectrolytes: PDADMA, PAH and QP4VP, respectively. All these gelators could form gel in toluene and their cmgc have been detected. The supramolecular structures and morphologies formed in the gel-phase have been characterized using TEM, AFM and SAXS. We found that the g2-PUA-PDADMA gelator demonstrated the lowest cmgc and the best networked ribbon-like structures in its gel-phase thus presented the strongest gel-forming ability among three DP gelators. The gel-forming ability of the g2-PUA-QP4VP gelator was worst, even worse than the parent g2-PUA gelator. The differences in the gel-forming ability and supramolecular morphology were originally due to the main-chain conformations of the three polyelectrolytes, which would further affect the fact-to-fact stacking of the g2-PUA dendrons in supramolecular morphology. The most ordered and rigid conformation in PDADMA polyelectrolyte could promote the gelation of the g2-PUA-PDADMA gelator because the g2-PUA dendrons could be pre-ordered along the backbone. However, the relatively disorder conformation of other two polyelectrolytes could even hinder the process of forming a pre-ordered structure of the g2-PUA dendrons, therefore, decline their gel-forming ability to some extent. Such the understanding is meaningful in directing us to enhance the gel-forming ability through complexation of such the dendritic gelators with an appropriate polyelectrolyte.Experimental
Materials
Poly(4-vinylpyridine) (P4VP, typical![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w ≈ 60
w ≈ 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and Poly(diallyldimethylammonium chloride) (PDADMAC, 20 wt% in water, typical w = 1.13 × 105 g mol−1) were purchased from ALDRICH. Poly(allylamine hydrochloride) (PAH, w ≈ 60000 g mol−1) were purchased from Alfa Aesar. Toluene was distilled under CaH2 situation. Methyl iodide (99%) and other reagents were purchased from major chemical suppliers and used without further purification unless otherwise noted.
000 g mol−1) and Poly(diallyldimethylammonium chloride) (PDADMAC, 20 wt% in water, typical w = 1.13 × 105 g mol−1) were purchased from ALDRICH. Poly(allylamine hydrochloride) (PAH, w ≈ 60000 g mol−1) were purchased from Alfa Aesar. Toluene was distilled under CaH2 situation. Methyl iodide (99%) and other reagents were purchased from major chemical suppliers and used without further purification unless otherwise noted.
Technique of analysis
(i) Transmission electron microscopy (TEM) examination was performed using JEOL JEM2010 electron microscopy. The samples were prepared by slight touch of gels on carbon film-coated copper grids; (ii) A MultiModel atomic force microscopy (Digital Instrumental Nanoscope IV) (AFM) was used to visualize aggregates on a silicon wafer surface at room temperature. The samples were prepared by dropping a drop of the solution on a freshly-cleaned silicon wafer surface. The solvent was removed in vacuo. All measurements were performed in tapping mode. (iii) 1H NMR spectra were recorded in d1-chloroform (CDCl3) on a NMR spectrometer (Varian UNITY Plus-400); (iv) The molecular mass of ionized g2-PUA was taken on a ESI-TOF mass spectrometer (Thermo Finnigan Co., Ltd. X7 ICP-MS); (v) Small angle X-ray scattering (SAXS) experiments were performed on Bruker AXS NANOSTAR using a monochromatic X-ray beam and a two-dimensional detector for scattering intensity recording. The distance between the samples and the detector is 270 mm. The plots of intensity vs. scattering wave vector, q, where θ is the scattering angle and λ = 1.54 Å for radiation beam, were produced by integrating the two-dimensional scattering patterns; (vi) The simulation on the conformation of polyelectrolytes was taken under the MMFF94 force field to obtain the state on minimized energy.Syntheses
:4, which proved a full quaternization in P4VP.
1H NMR spectra of g2-PUA-PAH (Fig. S4†) and g2-PUA-QP4VP (Fig. S5†) gave evidence for the complexes formation. From 1H NMR spectrum of g2-PUA-PAH in Fig. S4†, we could find the shift of signals (between 2.5 and 3.0 ppm) for the hydrogen on g2-PUA's apex, which proved the electrical density near the hydrogen grew more compact and gave an indirect evidence for the combination of g2-PUA and PAH. The reason for the weakness of PAH signals in g2-PUA-PAH should be the poor solubility of polyelectrolyte PAH in CDCl3 and embedding effect caused by dendrons around them. In Fig. S5† of g2-PUA-QP4VP we could also find the shift of signals on g2-PUA's apex (between 2.5 and 3.0 ppm) as g2-PUA-PAH, and fortunately we could see the signal of methyl on N atoms (4.35 ppm), which proved the existence of QP4VP in our product. But because of the embedding effect and poor solubility of QP4VP, other signals on aromatic groups (between 7.5 and 9.0 ppm) were still unclear. This phenomenon proved that the poor solubility of polyelectrolyte made it lack of their signal in 1H NMR spectra. But the precipitation from transparent solution, and the green color in g2-PUA-QP4VP solid both prove that the complexation between g2-PUA dendrons and polyelectrolytes occurred.
Preparation of gels
Gels of all dendronized polymer gelators and g2-PUA gelator were prepared following a facile method. The gelators were weighed in a clean transparent transform bottle, and a volume of redistilled toluene was added in to get the targeted concentration. Then the bottle was sealed with Teflon tape. The mixture was sonicated for 5 min and then heated at 70 °C for 5 min to get clear solution. And then the bottle was left standing still at 15 °C for a period of time until gel formed.Simulation
The chain conformation simulation of the three polyelectrolytes was performed using commercially available software Chem3D®. Under the MMFF94 force field the chain conformations with minimized energy were obtained from 30 repeating units of the polyelectrolytes in the condition of ignoring the effect of counter ions.Acknowledgements
This work was supported by the National Science Foundation of China (NSFC Grant No. 20734001) and State Key Laboratory of Polymer Physics and Chemistry.Notes and references
- I. M. Hamley, Introduction to Soft Matter, Synthetic and Biological Self-Assembling Materials, John Wiley & Sons, Chichester, 2007 Search PubMed.
- P. J. Flory, Faraday Discuss. Chem. Soc., 1974, 57, 7–18 RSC.
- Physical Properties of Polymeric Gels, ed. J. P. C. Addad, Wiley, New York, 1996 Search PubMed.
- For reviews on polymer gels, see: (a) A. Suzuki, Adv. Polym. Sci., 1993, 110, 201–240; (b) A. Keller, Faraday Discuss., 1995, 101, 1–49 RSC; (c) M. Shibayama, Macromol. Chem. Phys., 1998, 199, 1–30 CrossRef CAS; (d) Y. Osada and J.-P. Gong, Adv. Mater., 1998, 10, 827–837 CrossRef CAS; (e) P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1–11 CrossRef CAS; (f) M. K. Joo, M. H. Park, B. G. Choi and B. Jeong, J. Mater. Chem., 2009, 19, 5891–5905 RSC; (g) M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455–463 RSC.
- (a) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1870–1879; (b) B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS; (c) C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC; (d) S. Chaterjia, I. K. Kwonb and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS.
- (a) Low Molecular Mass Gelators. Design, Self-Assembly, Function, ed. F. Fages, Top. Current Chem., 2005, vol. 256 Search PubMed; (b) Molecular Gels. Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006 Search PubMed.
- For reviews on LMM gels, see: (a) P. Terech and G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef; (b) J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef; (c) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS; (d) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (e) M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS; (f) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS.
- Thematic issue on LMM gelators: Langmuir, 2009, 25, 8369–8840.
- (a) M. de Loos, J. van Esch, I. Stokroos, R. M. Kellogg and B. L. Feringa, J. Am. Chem. Soc., 1997, 119, 12675–12676 CrossRef CAS; (b) S. Yagai, T. Iwashima, T. Karatsu and A. Kitamaura, Chem. Commun., 2004, 1114–1115 RSC; (c) Y. Zhang, W. Huang, Y. Zhou and D. Yan, Chem. Commun., 2007, 2587–2589 RSC; (d) Y. Li, T. Wang and M. Liu, Soft Matter, 2007, 3, 1312–1317 RSC.
- (a) X.-Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871–3873 RSC; (b) C. Wang, D. Zhang and D. Zhu, Langmuir, 2007, 23, 1478–1482 CrossRef CAS; (c) X.-Q. Li, X. Zhang, S. Ghosh and F. Würthner, Chem.–Eur. J., 2008, 14, 8074–8078 CrossRef CAS.
- (a) U. Maitra, P. V. Kumar, N. Chandra, L. J. D'Souza, M. D. Prasanna and A. R. Raju, Chem. Commun., 1999, 595–596 RSC; (b) P. Babu, N. M. Sangeetha, P. Vijaykumar, U. Maitra, K. Rissanen and A. R. Raju, Chem.–Eur. J., 2003, 9, 1922–1932 CrossRef CAS.
- For reviews on supramolecular polymer gels, see: (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS; (b) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS; (c) J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS; (d) W. Weng, Z. Li, A. M. Jamieson and S. J. Rowan, Macromolecules, 2009, 42, 236–246 CrossRef CAS.
- (a) R. V. Ulijn, N. Bibi, V. Jayawarna, P. D. Thornton, S. J. Todd, R. J. Mart, A. M. Smith and J. E. Gough, Mater. Today, 2007, 10, 40–48 CrossRef CAS; (b) A. Vintiloiu and J.-C. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef CAS; (c) G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437–442 Search PubMed.
- For a review on self-assembled structure of dendrons, dendrimers and dendronized polymers, see: (a) F. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681–1712 CrossRef CAS; (b) G. R. Newkome, E. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689–1746 CrossRef CAS; (c) A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef CAS; (d) S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3867 CrossRef CAS; (e) B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS.
- (a) W.-D. Jang, D.-L. Jiang and T. Aida, J. Am. Chem. Soc., 2000, 122, 3232–3233 CrossRef CAS; (b) C. Kim, K. T. Kim and Y. Chang, J. Am. Chem. Soc., 2001, 123, 5586–5587 CrossRef CAS; (c) W.-D. Jang and T. Aida, Macromolecules, 2003, 36, 8461–8469 CrossRef CAS; (d) C. Kim, S. J. Lee, I. H. Lee and K. T. Kim, Chem. Mater., 2003, 15, 3638–3642 CrossRef CAS; (e) Y. Ji, Y.-F. Luo, X.-R. Jia, E.-Q. Chen, Y. Huang, C. Ye, B.-B. Wang, Q.-F. Zhou and Y. Wei, Angew. Chem., Int. Ed., 2005, 44, 6025–6029 CrossRef CAS; (f) H.-F. Chow and J. Zhang, Chem.–Eur. J., 2005, 11, 5817–5831 CrossRef CAS; (g) Y. Ji, G.-C. Kuang, X.-R. Jia, E.-Q. Chen, B.-B. Wang, W.-S. Li, Y. Wei and J. Lei, Chem. Commun., 2007, 4233–4235 RSC; (h) G.-C. Kuang, Y. Ji, X.-R. Jia, Y. Li, E.-Q. Chen and Y. Wei, Chem. Mater., 2008, 20, 4173–4175 CrossRef CAS.
- (a) A. R. Hirst, D. K. Smith, M. C. Feiters, H. P. M. Geurts and A. C. Wright, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef CAS; (b) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Chem.–Eur. J., 2004, 10, 5901–5910 CrossRef CAS; (c) H. S. Ko, C. Park, S. M. Lee, H. H. Song and C. Kim, Chem. Mater., 2004, 16, 3872–3876 CrossRef CAS; (d) B. Huang, A. R. Hirst, D. K. Smith, V. Castelletto and I. W. Hamley, J. Am. Chem. Soc., 2005, 127, 7130–7139 CrossRef CAS; (e) H.-J. Jeon, M. K. Kang, C. Park, K. T. Kim, J. Y. Chang, C. Kim and H. H. Song, Langmuir, 2007, 23, 13109–13116 CrossRef CAS; (f) V. Percec, M. Peterca, M. E. Yurchenko, J. G. Rudick and P. A. Heiney, Chem.–Eur. J., 2008, 14, 909–918 CrossRef CAS; (g) A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS; (h) G. Kuang, Y. Ji, X. Jia, E. Chen, M. Gao, J. Yeh and Y. Wei, Chem. Mater., 2009, 21, 456–462 CrossRef CAS; (i) P. Xue, R. Lu, X. Yang, L. Zhao, D. Xu, Y. Liu, H. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem.–Eur. J., 2009, 15, 9824–9835 CrossRef CAS.
- (a) M. Yang, Z. Zhang, F. Yuan, W. Wang, S. Hess, K. Lienkamp, I. Lieberwirth and G. Wegner, Chem.–Eur. J., 2008, 14, 3330–3337 CrossRef CAS; (b) Y. Feng, Z.-T. Liu, J. Liu, Y.-M. He, Q.-Y. Zheng and Q.-H. Fan, J. Am. Chem. Soc., 2009, 131, 7950–7951 CrossRef CAS; (c) M. Seo, J. H. Kim, J. Kim, N. Park, J. Park and S. Y. Kim, Chem.–Eur. J., 2010, 16, 2427–2441 CrossRef CAS.
- (a) E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2001, 123, 4105–4106 CrossRef; (b) K. T. Kim, C. Park, C. Kim, M. A. Winnik and I. Manners, Chem. Commun., 2006, 1372–1374 RSC; (c) E. R. Zubarev, E. D. Sone and S. I. Stupp, Chem.–Eur. J., 2006, 12, 7313–7327 CrossRef CAS.
- (a) M. Yoshida, Z. M. Fresco, S. Ohnishi and J. M. J. Fréchet, Macromolecules, 2005, 38, 334–344 CrossRef CAS; (b) C. Park, K. S. Choi, Y. Song, H.-J. Jeon, H. H. Song, J. Y. Chang and C. Kim, Langmuir, 2006, 22, 3812–3817 CrossRef CAS.
- (a) N. Canilho, E. Kasëmi, R. Mezzenga and A. D. Schlüter, J. Am. Chem. Soc., 2006, 128, 13998–13999 CrossRef CAS; (b) X. Zhu, U. Beginn, M. Möller, R. I. Gearba, D. V. Anokhin and D. A. Ivanov, J. Am. Chem. Soc., 2006, 128, 16928–16937 CrossRef CAS; (c) Z. Cheng, B. Ren, H. Shan, X. Liu and Z. Tong, Macromolecules, 2008, 41, 2656–2662 CrossRef CAS; (d) X. Zhang, Y. Wang, W. Wang, S. Bolisetty, Y. Lu and M. Ballauff, Langmuir, 2009, 25, 2075–2080 CrossRef CAS; (e) X.-J. Zhang, F. Yuan, Y.-L. Wang, Z.-J. Zhang and W. Wang, Chin. J. Polym. Sci., 2010, 28, 395–404 CAS.
- Z. Zhang, M. Yang, X. Zhang, L. Zhang, B. Liu, P. Zheng and W. Wang, Chem. Eur. J., 2009, 15, 2352–2361 CrossRef CAS.
- Because of kinetic feature of gelation process, the minimum gelation concentration (cmgc) is a function of gelation time. Normally, cmgc decreases with increasing gelation time. In this study, cmgc values of the three dendronized polymer gelators were determined at a constant gelation time of one day. For the g2-PUA gelator a two-day gelation time was used for determination of its cmgc value because of its low gelation ability.
- (a) K. W. Mattison, P. L. Dubin and I. J. Brittain, J. Phys. Chem. B, 1998, 102, 3830–3836 CrossRef CAS; (b) A. B. Kayitmazer, D. Shaw and P. L. Dubin, Macromolecules, 2005, 38, 5198–5204 CrossRef CAS.
- C. Burger, S. Zhou and B. Chu, in Handbook of Polyelectrolytes and Their Applications, ed. S. K. Tripathy, J. Kumar and H. S. Nalwa, American Scientific Publishers, Stevenson Ranch, California, 2002, vol 3, pp. 125–141 Search PubMed.
- C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- M. Yang, W. Wang, I. Lieberwirth and G. Wegner, J. Am. Chem. Soc., 2009, 131, 6283–6292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of n-hexadecanol, g2-PUA dendron, ionized g2-PUA, QP4VP, PAH, g2-PUA-PAH and g2-PUA-QP4VP. ESI-MS spectrum of ionized g2-PUA. 3D AFM height image of the dried-samples of the g2-PUA-PDADMA, g2-PUA-PAH and g2-PUA-QP4VP gels. See DOI: 10.1039/c0nj00371a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |