DOI:
10.1039/C0NJ00583E
(Paper)
New J. Chem., 2011,
35, 149-155
A recyclable polymer-supported ruthenium catalyst for the oxidative degradation of bisphenol A in water using hydrogen peroxide†
Received
(in Montpellier, France)
25th July 2010
, Accepted 19th August 2010
First published on 8th October 2010
Abstract
A polypyridyl ruthenium(II) complex, cis-[RuII(2,9-Me2phen)2(H2O)2]2+, has been adsorbed onto the cation-exchange resins Dowex-50W and Chelex-100. The potential use of the supported ruthenium(II) complex as catalyst for the oxidative degradation of organic pollutants in water has been investigated using bisphenol A, an emerging endocrine disruptor, as substrate; and the environmentally friendly H2O2 as oxidant. These solid-supported catalysts are found to be efficient for the degradation of bisphenol A in aqueous solution by H2O2 under ambient conditions. The intermediates and products formed during the oxidative degradation of bisphenol A by these catalytic systems have been identified and a mechanism is proposed. The supported catalysts are easily recovered by simple filtration and display no loss of activity when recycled.
Introduction
A variety of ruthenium complexes, especially those bearing polypyridyl or macrocyclic tertiary amine ligands, are active catalysts for the oxidation of various organic compounds including alkanes, alkenes, alkynes and alcohols; using terminal oxidants such as Ce(IV), tert-butyl hydroperoxide (TBHP), hydrogen peroxide or O2.1–12 Although these ruthenium complexes are among the most active catalysts for selective organic oxidation, their use as catalysts for the oxidative degradation of organic pollutants in water has not been explored. The amount of organic pollutants in wastewaters discharged by various industries is increasing every year and is causing serious global environmental problems. These pollutants include a wide range of persistent organic chemicals, such as pharmaceuticals and endocrine-disrupting compounds. We are now faced with the challenge of removing these compounds from their effluent before they are discharged. Although catalytic oxidation processes using Fenton (Fe2+ + H2O2) and photo-Fenton (Fe2+ + H2O2 + UV) reagents have been developed for organic pollutants removal from wastewaters, these methods are rather inefficient.13–16 Moreover, the Fenton reaction is only effective at low pH (<4), since at higher pH Fe(OH)3 precipitate will form which is inactive.16,17Fenton reaction also results in inevitable flocs formation which requires time consuming further treatments. In general, ruthenium complexes are much more stable and active oxidation catalysts than iron complexes, it is anticipated that they should also be useful catalysts for the oxidative degradation of organic pollutants in water. Although ruthenium is more toxic and expensive than iron, if it can be made in the form of a robust solid-supported catalyst that can be readily recyclable without leaching into the environment, then its use is still a feasible process. We report herein the use of a ruthenium(II) complex, cis-[RuII(2,9-Me2phen)2(H2O)2](PF6)2 (2,9-Me2phen = 2,9-dimethyl-1,10-phenanthroline), that is supported on a cation-exchange resin (Dowex-50W or Chelex-100), as efficient catalyst for the oxidative degradation of bisphenol A (2,2-bis(4-hydroxyphenyl)propane), using hydrogen peroxide as the oxidant under ambient conditions at various pH. Bisphenol A (structure f in Table 1) is a known endocrine disruptor which has been widely used for the production of polycarbonate (PC) and epoxy resins used in food containers.18,19 Its concentration in wastewaters is increasing worldwide.20 This is the first report on the use of a solid-supported ruthenium catalyst for environmental remediation.
Table 1 List of major intermediates (a–e) and main fragments (m/z) obtained by LC-MS.
| Peak |
Retention time/min |
m/z (amu) |
Molecular weight |
MS/MS fragments (relative abundance) |
Proposed structure |
|
a
|
5.5 |
163 |
164 |
163(100) |
|
| 107(64) |
|
b
|
7.2 |
163 |
164 |
163(100) |

|
| 107(64) |
|
c
|
9.6 |
135 |
136 |
135(45) |
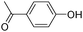
|
| 120(27) |
| 92(100) |
|
d
|
12.0 |
257 |
258 |
257(100) |
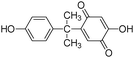
|
| 242(18) |
| 229(97) |
| 214(50) |
|
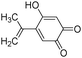
e
|
14.3 |
241 |
242 |
241(44) |
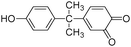
|
| 226(100) |
|
f
|
16.3 |
227 |
228 |
227(100) |
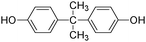
|
| 212(63) |
| 133(20) |
Results and discussion
Preparation and characterization of supported catalysts
Upon adding Dowex-50W or Chelex-100 to a red solution of cis-[Ru(2,9-Me2phen)2(OH2)2]2+ (RuIIII) in water, the colour of the solution was rapidly discharged while the resin turned red, indicating adsorption of the ruthenium(II) complex onto the resin. Analysis of the ruthenium content by ICP-AES after leaching with 6 M HCl indicates that the maximum adsorption capacity of the resins are 8.2 μmol RuIIII/g Dowex-50W and 6.9 μmol RuIIII/g Chelex-100. Complete adsorption occurs if the amount of RuIIII used is less than these values. No RuIIII was detected in the solution after continuous stirring of a RuIIII-Dowex suspension in water or in 1 M HNO3 for 4 h. On the other hand, although RuIIII-Chelex is stable in water or 0.1 M HNO3, complete leaching of RuIIII occurs in 1.0 M HNO3, as monitored by UV/Vis spectrophotometry (RuIIII: λmax = 496 nm, ε = 7.1 × 103 mol−1 dm3 cm−1).
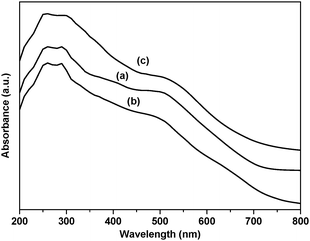
The diffuse reflectance spectrum of RuIIII-Dowex shows a broad peak with λmax at around 500 nm (Fig. 1), which is similar to that of RuIIII in water (496 nm). On the other hand, the diffuse reflectance spectrum of RuIIII-Chelex shows λmax at around 520 nm, slightly red-shifted compared with the unsupported RuII complex.
Oxidative degradation of bisphenol A
The potential of the polymer-supported ruthenium complex as catalyst for oxidative degradation of organic pollutants in water was investigated using bisphenol A (BPA) as the substrate and the environmentally friendly hydrogen peroxide as oxidizing agent. Upon adding RuIIII-Dowex or RuIIII-Chelex to a solution of bisphenol A in water containing H2O2, the concentration of bisphenol A decreased with time, as monitored by LC and LC/MS (Fig. 2). A 0.50 mM solution of bisphenol A in water can be completely degraded by 25 mM of H2O2 using the unsupported RuIIII, RuIIII-Dowex or RuIIII-Chelex. Theoretically 36 moles of H2O2 are required to completely oxidize 1 mole of BPA to CO2 and H2O. As expected, RuIIII has the highest activity, while RuIIII-Chelex is the least active catalyst, although it has a faster initial rate than RuIIII-Dowex. RuCl3·xH2O, which is a common ruthenium complex, is much less effective. No degradation of BPA occurred in the absence of ruthenium catalysts. Notably the solutions containing the supported catalysts remained colourless throughout the catalytic oxidation, the absorbances at 496 nm (λmax of RuII) were <0.005, indicating that [RuIIII] in the solutions were <10−7 M. In an independent experiment, no oxidation of bisphenol A was found to occur when a 1 × 10−7 M RuIIII was used as catalyst. These results strongly suggest that the active catalyst is the supported one.
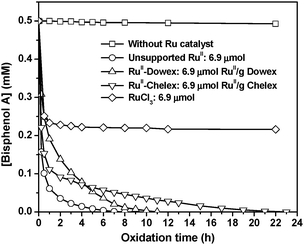 |
| | Fig. 2 Time course for the ruthenium catalyzed degradation of BPA (0.50 mM) by H2O2 (25 mM) in water (50 mL). T = 25 °C. | |
Since RuIIII-Dowex is more active than RuIIII-Chelex, it was selected for further investigations. Fig. 3 shows the effect of H2O2 concentration on the degradation of bisphenol A. As expected, lowering the H2O2 concentration leads to a decrease in reaction rate. Also, complete degradation of bisphenol A is not achieved using 5 mM or 10 mM H2O2.
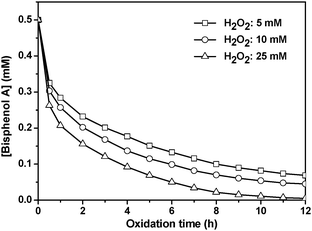 |
| | Fig. 3 Effect of H2O2 concentration on degradation of BPA. RuIIII-Dowex (8.2 μmol RuIIII/g Dowex), 1.0 g; bisphenol A, (0.50 mM) in 50 mL water. T = 25 °C. | |
The decrease in [BPA] at various [H2O2] was found to obey first-order kinetics. Plots of ln(C0/C) versus time t are linear (C0 = initial concentration, C = concentration at time t), and the slopes give the pseudo-first-order rate constants kobs. The plot of 1/kobsversus 1/[H2O2] is linear (Fig. 4), which is consistent with equilibrium formation of a precursor complex between RuIIII-Dowex and H2O2 prior to the oxidation of BPA, as shown in eqn (1) and (2). The rate law is shown in eqn (3); k and K are found to be (3.24 ± 0.45) × 10−1s−1 and (430 ± 10) M−1, respectively at 25 °C.
| | 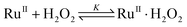 | (1) |
| | 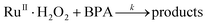 | (2) |
| | 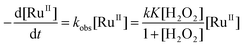 | (3) |
The effect of the amount of
catalyst on the degradation of
bisphenol A is illustrated in
Fig. 5.
![Plot of 1/kobsversus 1/[H2O2] for the RuIIII-Dowex catalyzed oxidative degradation of BPA by H2O2. Slope = (7.12 ± 0.88) × 10−3, y-intercept = 3.09 ± 0.11, r = 0.9924. T = 25 °C.](/image/article/2011/NJ/c0nj00583e/c0nj00583e-f4.gif) |
| | Fig. 4 Plot of 1/kobsversus 1/[H2O2] for the RuIIII-Dowex catalyzed oxidative degradation of BPA by H2O2. Slope = (7.12 ± 0.88) × 10−3, y-intercept = 3.09 ± 0.11, r = 0.9924. T = 25 °C. | |
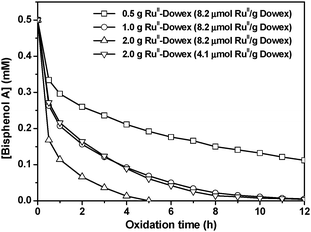 |
| | Fig. 5 Effect of the amount of RuIIII-Dowexcatalyst on the oxidative degradation of BPA. H2O2, 25 mM; bisphenol A, 0.50 mM in 50 mL water. T = 25 °C. | |
As expected, the rate of oxidation of bisphenol A increases with the amount of catalyst (2 g > 1 g > 0.5 g). The decrease in [BPA] obeys first-order-kinetics, and the pseduo-first-order rate constants are found to increase linearly with the amount of catalyst (Fig. 6). Complete degradation of bisphenol A was not achieved when 0.5 g of catalyst was used. On the other hand, when 2 g of catalyst with half the ruthenium loading (4.1 μmol RuIIII/g) was used, the rate is similar to using 1 g of catalyst with ruthenium loading of 8.2 μmol RuIIII/g, although the former catalyst should have a higher surface area.
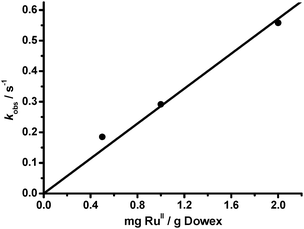 |
| | Fig. 6 Plot of kobsversus amount of RuIIII-Dowex in the oxidative degradation of BPA by H2O2. Slope = (2.86 ± 0.14) × 10−1 g−1s−1, r = 0.9986. T = 25 °C. | |
The catalytic activity of RuIIII-Dowex was also investigated at various pH. As shown in Fig. 7, RuIIII-Dowex is an efficient catalyst at pH 4–9, which is the range of pH found in most natural and polluted waters. The activity follows the order pH 8 > 9 > 7 > 4. In general, the catalyst exhibits higher activity at high pH. In this regard, this system is better than the Fenton reagent, which is ineffective at pH > 4 due to the precipitation of Fe(OH)3.17
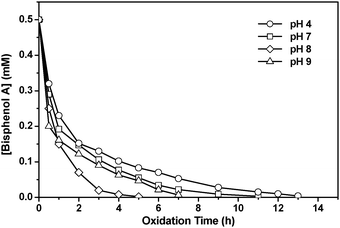 |
| | Fig. 7 Oxidative degradation of BPA (0.5 mM) catalyzed by RuIIII-Dowex at various pH. H2O2, 25 mM; RuIIII-Dowex, 1.0 g (8.2 μmol RuIIII); 50 mL water. T = 25 °C. | |
Recycling of the polymer-supported RuIIIIcatalyst
After catalytic reaction, the solution was centrifuged. The RuIIII-Resin was collected, washed with deionized water and then used again for oxidation degradation of bisphenol A under the same conditions as before. It was found that there was virtually no loss of activity after three successive catalyst recyclings. The ruthenium content of the resin after three recyclings remained the same as analyzed by ICP. There was also no decomposition nor leaching of the ruthenium complex into the solution, as monitored by UV-Vis spectrophotometry. Moreover, the diffuse reflectance spectra of RuII-resin before and after catalytic oxidation are the same. In addition, little or no decomposition of RuII was also found to occur in homogeneous catalytic oxidation, as illustrated in Fig. 8. The UV-Vis spectra of RuII before and after catalytic oxidation are very similar, the difference in the UV region is probably due to the presence of oxidized organic products.
![UV-Vis spectra of RuIIII in homogeneous catalytic oxidation. (a) Before adding H2O2, [RuIIII] = 0.07 mM, [BPA] = 0.5 mM. (b) Immediately after adding H2O2, [H2O2] = 25 mM, (c) After 5 h, excess Na2SO3 added to remove H2O2.](/image/article/2011/NJ/c0nj00583e/c0nj00583e-f8.gif) |
| | Fig. 8
UV-Vis spectra of RuIIII in homogeneous catalytic oxidation. (a) Before adding H2O2, [RuIIII] = 0.07 mM, [BPA] = 0.5 mM. (b) Immediately after adding H2O2, [H2O2] = 25 mM, (c) After 5 h, excess Na2SO3 added to remove H2O2. | |
Analysis of the intermediates and products formed in the oxidative degradation of bisphenol A
The intermediates formed during the degradation of bisphenol A were identified by LC/MS and LC/MS/MS experiments. Fig. 9 shows the total ions chromatogram (TIC) from LC/MS for the intermediates formed during bisphenol A degradation by RuIIII-Dowex after 7 h. Five major intermediates were identified, as listed in Table 1.
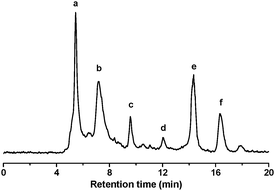 |
| | Fig. 9 Liquid chromatogram of the reaction mixture after 7 h at 23 °C. BPA, 0.5 mM; H2O2, 25 mM; RuIIII-Dowex, 1.0 g (8.2 μmol RuIIII); 50 mL water. | |
In the liquid chromatogram, the peak f is bisphenol A with a m/z of 227. The m/z value for each peak corresponds to the [M–H]− ion that arises from deprotonation of a hydroxyl group. Since a reverse-phase LC column and a polar mobile phase were used, the products were eluted in decreasing order of polarity. Each peak was identified by interpretation of the MS/MS spectra. Similar intermediates have also been reported for the degradation of bisphenol A using Fenton's reagent15 and TiO2 photocatalyst.21 The compounds in peak a and b have the same molecular weight and fragment ions, indicating that they are closely related isomers. The compound in peak a should be more polar, hence it is assigned as the ortho-quinone derivative, while that in peak b is assigned as the para-quinone derivative.
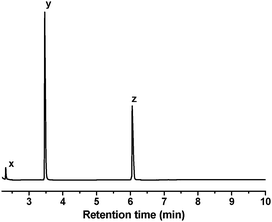
Although these aromatic intermediates could still have endocrine disrupting effects, they all eventually disappeared with time. In the LC of the reaction mixture after 24 h (Fig. 10), there is only one major peak. The main species in this peak was identified as butenedioic acid (HCOOCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOH). GC-MS was also used to identify any volatile products not detected by LC-MS. Direct injection of the reaction mixture into GC-MS gave only one major peak which consists of N2, O2 and CO2 with relative abundance of 69, 100 and 75, respectively. The amount of carbon dioxide detected is far greater than the background level in the atmosphere, indicating that it is derived from oxidative degradation of bisphenol A. Additional CO2 could have escaped into the atmosphere since the oxidation reactions were done in the open air. The reaction solution was also extracted with ether (2 × 1 mL) and then analyzed by GC-MS (Fig. 11). In this case three major peaks were observed, which were identified as formic acid (x), oxalic acid (y) and acetic acid (z).
CHCOOH). GC-MS was also used to identify any volatile products not detected by LC-MS. Direct injection of the reaction mixture into GC-MS gave only one major peak which consists of N2, O2 and CO2 with relative abundance of 69, 100 and 75, respectively. The amount of carbon dioxide detected is far greater than the background level in the atmosphere, indicating that it is derived from oxidative degradation of bisphenol A. Additional CO2 could have escaped into the atmosphere since the oxidation reactions were done in the open air. The reaction solution was also extracted with ether (2 × 1 mL) and then analyzed by GC-MS (Fig. 11). In this case three major peaks were observed, which were identified as formic acid (x), oxalic acid (y) and acetic acid (z).
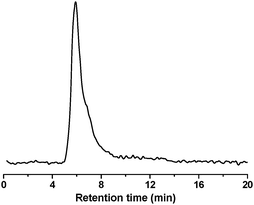 |
| | Fig. 10 Liquid chromatogram of the mixture containing BPA, H2O2 and RuIIII-Dowexcatalyst after 24 h reaction. | |
Proposed degradation pathways of bisphenol A
Based on the observed intermediates and products, it is evident that the degradation pathways involve a Fenton mechanism which produces hydroxyl radicals (˙OH) according to eqn (4).| | | RuIIII + H2O2 → RuIIIIIIOH + ˙OH | (4) |
The proposed oxidative degradation pathways for bisphenol A are shown in Fig. 12. The first step is the addition of ˙OH to an aromatic ring of bisphenol A to give I. The formation of hydroxylated derivatives by the reaction of hydroxyl radicals with aromatic rings is well-known.22–25 The radical species I may then undergo hydrogen-atom abstraction (HAT) by RuIIIIII–OH to give the dihydroxylated derivative II, which can be further oxidized to give the trihydroxylated derivative III by the same mechanism. II and III are then oxidized viaHAT, first by ˙OH and then by RuIIIIIIOH, to give the quinones d and e. These phenolic and quinone derivatives of bisphenol A then undergo oxidative C–C bond cleavage by ˙OH to give benzophenone, substituted benzophenones and substituted phenols; which undergo further oxidation and ring degradation by ˙OH to finally give butenedioic acid, formic acid, acetic acid, oxalic acid and carbon dioxide.
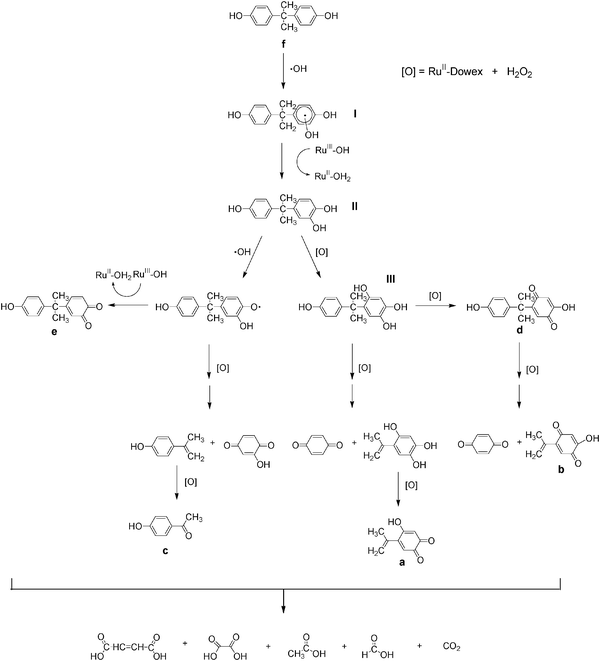 |
| | Fig. 12 Proposed oxidative degradation pathways of BPA by RuIIII-Dowex/H2O2. | |
This catalytic system is reasonably efficient. In our studies we found that 50 equivalents of H2O2 are required for complete disappearance of bisphenol A. Complete conversion to CO2 and H2O would require 36 equivalents, whereas degradation to butenedioic acid requires 14 equivalents. Since a mixture of aliphatic acids and CO2 were produced, we estimate that around 25 equivalents of H2O2 are used for the degradation of bisphenol A, the remaining H2O2 probably decomposes to H2O and O2.
RuIIII
versus
FeII/FeIII as catalyst
The highly catalytic activity of polypyridyl ruthenium complexes such as [RuII(2,9-Me2phen)2(H2O)2]2+ compared to simple iron(II)/(III) salts may arise from the following factors. First, because of strong π-backbonding between Ru(II) and polypyridyl ligands, the geometry of Ru(II) is very similar to that of Ru(III), hence inter-conversion between the two oxidation states is rapid. For example, the self-exchange rate of [Ru(bpy)3]2+/3+ is 2 × 109 M−1s−1,26 while that of [Fe(OH2)6]2+/3+ is 4 M−1s−1.27 Hence the redox reaction between RuIIII and H2O2 to generate ˙OH is expected to be fast. On the other hand, Ru(III) aqua and hydroxo complexes bearing polypyridyl ligands are strong oxidants that can readily oxidize organic substrates via one-electron transfer or HAT.5,28 This means RuIIIIII–OH can be readily converted back to RuIIII which can then react with H2O2 to generate more ˙OH. Moreover polypyridyl ruthenium(II) complexes are usually very stable at room temperature in aqueous solutions.5
Conclusions
The oxidative degradation of bisphenol A by H2O2 catalyzed by a polypyridyl ruthenium(II) complex, cis-[RuII(2,9-Me2phen)2(H2O)2]2+, that is supported on the cation-exchange resin Dowex-50W or Chelex-100, has been studied. The supported catalyst can decompose bisphenol A in aqueous solution with H2O2 as oxidant to give carbon dioxide and biodegradable aliphatic acids. The RuIIII-Resin is a much more active catalyst than simple iron(II)/(III) salts. Moreover it is active at a wide range of pH values, whereas the Fenton reagent can only function at pH < 4. The RuIIII-Resincatalyst can be readily reused without loss of activity. In conclusion, RuIIII-Dowex/H2O2 is potentially a useful catalytic system for the degradation of organic pollutants in water at a wide range of pH.
Experimental
Materials
Bisphenol A, H2O2 (35% by weight) and 2,9-dimethyl-1,10-phenanthroline (2,9-Me2phen) were obtained from Sigma-Aldrich. Chelex-100 resin (sodium form, with iminodiacetate functional group) with 1% cross-linking and of 50–100 dry mesh size was also purchased from Sigma-Aldrich. Dowex-50W cation-exchange resin (with sulfonate functional group) with 8% cross-linking and of 50–100 dry mesh size was obtained from Dow Chemical Company. The resins were first washed with 1.0 M HCl, then with doubly distilled deionized water and dried at 100 °C for 2 h before use. Acetonitrile (HPLC grade) was obtained from Lab-Scan. All other chemicals were of analytical grade and were used without further purification. cis-[RuII(2,9-Me2phen)2(H2O)2](PF6)2 was prepared by a literature procedure.8 Found: C, 39.71; H, 3.49; N, 6.54. Calc. for C28F12H28N4O2P2Ru: C, 39.86; H, 3.35; N, 6.64. λmax/nm (ε, dm3 mol−1 cm−1) in water: 496 (1.02 × 104).
Immobilization of ruthenium catalyst onto resins
Typically 1 g of resin (Dowex-50W or Chelex-100) was added to a red solution of cis-[RuII(2,9-Me2phen)2(H2O)2](PF6)2 (RuIIII, 3–8 mg) in water (5 ml) at room temperature with continuous stirring. The colour of the resin gradually turned red, indicating adsorption of the ruthenium complex onto its surface. After 1 h, the solution was filtered and the RuIIII-Resin was washed with water and dried in vacuo.
Characterization of the supported catalysts
Diffuse reflectance UV-vis spectra of the RuIIII-Resins before and after catalytic oxidation were obtained using a Perkin Elmer Lambda 900 spectrophotometer. Since RuIIII-Resins after catalytic reaction were turned from pellet into powder form due to the stirring process, the RuIIII-Resins before catalysis were also ground into powder form for direct comparison.
The amount of catalyst adsorbed onto the resin was determined by adding 6 M HCl to RuIIII-Resin at room temperature with stirring for 1 h. The resin turned back to its original color and the HCl solution became red. The ruthenium content in the HCl solution was determined by a Perkin-Elmer Plasma 1000 inductively-coupled plasma atomic emission spectrometer (ICP-AES).
Oxidative degradation of bisphenol A
RuIIII-Resin was added to an aqueous solution containing bisphenol A and hydrogen peroxide, and the mixture was stirred at 500 rpm at 25 °C. Aliquots were withdrawn at various time intervals and centrifuged. Analysis of bisphenol A, intermediates and products were carried out by an Agilent 1200 series liquid chromatograph equipped with an Agilent C18 column, a UV detector and a API 2000 mass spectrometer (electrospray ionization). The mass spectrometer was operated at the anionic mode in the m/z range 50–500 for LC/MS and 50–300 for LC/MS/MS. The mobile phase was acetonitrile–water (55/45, v/v) with a flow rate of 200 μL min−1. Final degradation products were also identified by GC-MS using an Agilent 6890 GC equipped with a DB-FFAP capillary column and an Agilent 5975 MSD mass selective detector.
Acknowledgements
The work described in this paper was supported by grants from the University Grants Committee of Hong Kong Special Administrative Region (CityU 2/06C, AoE/P-04/04). H.L. is also grateful for financial support provided by the Outstanding Youth Fund (No. 20525416) and National Natural Science Foundation of China (No. 20874094).
Notes and references
-
T. V. Lee, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and S. V. Ley, Pergamon, Oxford, 1991, vol. 7, p. 291 Search PubMed.
- T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599 CrossRef CAS.
- S.-I. Murahashi, N. Komiya, Y. Oda, T. Kuwabara and T. Naota, J. Org. Chem., 2000, 65, 9186 CrossRef.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144 CrossRef CAS.
- T. J. Meyer and M. Y. V. Huynh, Inorg. Chem., 2003, 42, 8140 CrossRef CAS.
- T. Kojima, H. Matsuo and Y. Matsuda, Inorg. Chim. Acta, 2000, 300–302, 661 CrossRef CAS.
- Y. Hirai, T. Kojima, Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 5772 CrossRef CAS.
- A. S. Goldstein, R. H. Beer and R. S. Drago, J. Am. Chem. Soc., 1994, 116, 2424 CrossRef CAS.
- T. C. Lau, C. M. Che, W. O. Lee and C. K. Poon, J. Chem. Soc., Chem. Commun., 1988, 1406 RSC.
- C. M. Che, K. W. Cheng, M. C. W. Chan, T. C. Lau and C. K. Mak, J. Org. Chem., 2000, 65, 7996 CrossRef CAS.
- C. M. Che, W. P. Yip and W. Y. Yu, Chem.–Asian J., 2006, 1, 453–458 CrossRef CAS.
- W. P. Yip, C. M. Ho, N. Zhu, T. C. Lau and C. M. Che, Chem.–Asian J., 2008, 3, 70 CrossRef CAS.
- Y. B. Xie and X. Z. Li, J. Hazard. Mater., 2006, 138, 526–533 CrossRef CAS.
- R. A. Torres, F. Abdelmalek, E. Combet, C. Pétrier and C. Pulgarin, J. Hazard. Mater., 2007, 146, 546–551 CrossRef CAS.
- B. Gözmen, M. A. Oturan and N. Oturan O. Erbatur, Environ. Sci. Technol., 2003, 37, 3716–3723 CrossRef.
- D. Zhou, F. Wu, N. Deng and W. Xiang, Water Res., 2004, 38, 4107–4116 CrossRef CAS.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS.
- J. Sajiki and J. Yonekubo, Chemosphere, 2002, 46, 345–354 CrossRef CAS.
- J. Y. Hu, T. Aizawa and S. Ookubo, Environ. Sci. Technol., 2002, 36, 1980–1987 CrossRef CAS.
- G. M. Klecka, C. A. Staples, K. E. Clark, N. V. D. Hoeven, D. E. Thomas and S. G. Hentges, Environ. Sci. Technol., 2009, 43, 6145–6150 CrossRef CAS.
- S. Fukahori, H. Ichiura, T. Kitaoka and H. Tanaka, Appl. Catal., B, 2003, 46, 453 CrossRef CAS.
- R. F. Anderson, K. B. Patel and M. R. L. Stratford, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3177 RSC.
- G. W. Klein, K. Bhatia, V. Madhavan and R. H. Schuler, J. Phys. Chem., 1975, 79, 1763 CrossRef.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- C. Walling and R. A. Johnson, J. Am. Chem. Soc., 1975, 97, 363–367 CrossRef CAS.
- D. J. Szalda, D. H. Macartney and N. Sutin, Inorg. Chem., 1984, 23, 3473 CrossRef CAS.
- J. Silverman and R. W. Dodson, J. Phys. Chem., 1952, 56, 846 CrossRef.
- R. A. Binstead, M. E. McGuire, A. Dovletoglou, W. K. Seok, L. E. Roecker and T. J. Meyer, J. Am. Chem. Soc., 1992, 114, 173–186 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.