X-Ray crystallographic and spectroscopic properties of eight Schiff bases as evidence of the proton transfer reaction. Role of the intermolecular hydrogen bond†
Oscar
Domínguez
a,
Braulio
Rodríguez-Molina
a,
Mario
Rodríguez
b,
Armando
Ariza
a,
Norberto
Farfán
c and
Rosa
Santillan
*a
aDepartamento de Química, Centro de Investigación y de Estudios Avanzados del IPN, CINVESTAV, Apdo., Postal 14-740, México, D. F., 07000, México. E-mail: rsantill@cinvestav.mx; Fax: +52 555-747-3389; Tel: +52 555-747-3725
bCentro de Investigaciones en Óptica, CIO, Apdo., Postal 1-948, 37000 León Gto., México
cFacultad de Química, Departamento de Química Orgánica, Universidad Nacional Autónoma de México, México, D.F., 04510, México
First published on 11th October 2010
Abstract
A spectroscopic study of several ortho-hydroxy Schiff bases was carried out, and the corresponding crystal structures were analyzed in order to identify their characteristic hydrogen bonding patterns. The X-ray analysis showed that the enol (O–H⋯N) tautomer is the most stable in compounds 1–3 whereas the keto (N–H⋯O) form is preferred in compounds 4–7. The specific intermolecular O–H⋯O hydrogen bonding interactions that control the supramolecular arrangement of each tautomer are discussed. Additionally, a complete characterization of the polycrystalline samples was attained using solid-state NMR and IR experiments. Solution VT NMR and UV-visible experiments were also used to obtain valuable insights about the nature and stability of the tautomers.
Introduction
Schiff bases are important organic compounds of successful application in several areas, such as biological chemistry1 materials science2 and organic synthesis.3 A tautomeric equilibrium between the enol O–H⋯N and keto N–H⋯O forms is commonly present in derivatives of aromatic-o-hydroxyaldehydes. A reversible intramolecular proton transfer driven by electrostatic differences between the oxygen and nitrogen atoms of the salicylideneimine fragment promotes the delocalization of the aromatic π-electron system leading to a quinoid form, which is also related with its canonical zwitterionic form4 (Scheme 1).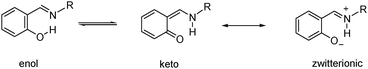 | ||
| Scheme 1 Tautomeric equilibrium for arylimine derivatives. | ||
Furthermore, it has been described that this tautomerism can be suitably controlled in the solid state by using light or temperature, depending on the photo- or thermochromic properties of the molecules,5 making these compounds potential candidates for optical switches and storage devices.6
Although extensive studies of several Schiff bases have been carried out, most of them are related with the synthesis and spectroscopic characterization of the tautomers,7 or merely focused on simple structural description of the crystal structures.8 Moreover, although the tautomeric equilibrum of the Schiff bases in solution has been widely explored,9 studies concerning the factors that determine the formation of the tautomers in the solid-state are scarce.10
A preliminary work by Ogawaet al. provided the first X-ray evidence of the effects of the intermolecular contacts on the stabilization of the keto tautomers in salicylideneimine derivatives.12 Subsequently, he showed that the proton transfer reaction in nonpolar solvents might be controlled by the aggregation state of the molecules at low temperatures13 (Scheme 2).
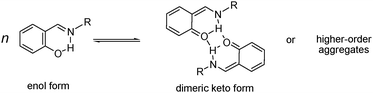 | ||
| Scheme 2 Intramolecular cyclic dimer that stabilizes keto tautomers in solution. | ||
In a previous paper, we reviewed the stabilizing effect of intermolecular contacts of keto forms of imine derivatives from salicylaldehyde and substituted anilines.11 In this work, we extended the investigation to eight ortho-hydroxy Schiff bases to unveil the influence (electronic and/or structural) of the substituents on the tautomeric structure from a crystallographic perspective (Scheme 3). In the selection of substituents were considered weak or good electron donors as tert-butyl and diethylamine groups, and electron withdrawing substituents such as, –NO2, and other groups like –Cl and –Br, to explore the acidity of the salicylidene fragment in the proton transfer process. Furthermore, large groups as diphenyl or t-butyl groups were introduced to evaluate the influence of the steric effect on the tautomerism.
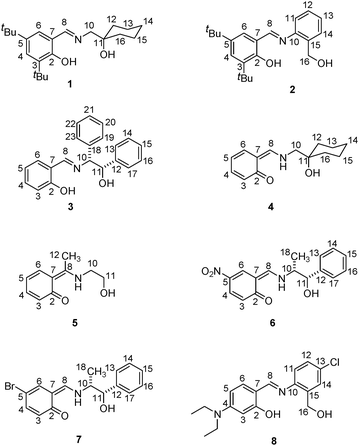 | ||
| Scheme 3 Molecular structures of the Schiff bases studied. Compounds are depicted as observed in X-ray diffraction. For compound 8 the enol tautomer is depicted. | ||
Besides the intramolecular hydrogen bonds present in the salicylidene fragment, the X-ray diffraction analysis revealed the existence of five specific intermolecular hydrogen-bonding patterns due to well aligned O–H⋯O hydrogen bonds: zigzag chains (compound 1, Scheme 4a), tetrameric rings (imine 2, Scheme 4b), linear chains (compounds 3–4, Scheme 4c), helical chains (derivative 5, Scheme 4d), cyclic dimeric rings (compounds 6–7, Scheme 4e) or more complex arrangements such as those observed in compound 8 (Fig. 5).
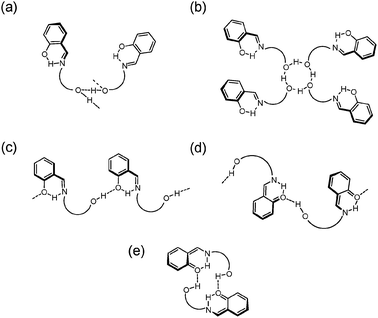 | ||
| Scheme 4 Intermolecular interactions observed in imine derivatives. | ||
Complementary UV-visible and solution NMR experiments supplied valuable insights about the tautomerization of these compounds and proved that the main attained form in solution is not directly related with that obtained in the solid-state, where the tautomeric stabilization is completely dependent on the prevailing contributions in the liquid phase (dipoles moments, solvation effects, molecular aggregates, etc.)10a,14 or in the solid phase (structural and electronic effects, hydrogen-bonding, etc.).15
Results and discussion
Characterization of compounds 1–8 in solution
Compounds 1–8 were prepared from the condensation reaction in methanol of different aminoalcohols and the corresponding salicylaldehyde. An important aspect of these compounds is the introduction of a complementary hydroxyl group, pertaining to the aminoalcohol unit labeled (O2–H2), in addition to the corresponding OH-salicylidene group marked as (O1–H1) that may confer additional stability to the supramolecular structures of all derivatives by establishing complementary interactions. In derivatives 2 and 8, the hydrogen-bonding interactions among neighboring molecules are additionally strengthened by minimization of the steric repulsions.15The use of absorption spectroscopy (UV-vis) for the identification of tautomeric species in solution16 has permitted to propose that the formation of intermolecular hydrogen-bonded aggregates at low temperature stabilizes the keto forms in nonpolar solvents13 (Scheme 2), even though the structure of these aggregates has not been clearly determined. In order to characterize the behavior of compounds 1–8 in solution, we measured the absorption spectra in different solvents at room temperature. Fig. 1 contains the spectra of compounds 4 and 5.
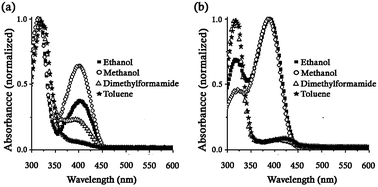 | ||
| Fig. 1 UV-Vis spectra of compounds (a) 4 and (b) 5 in solvents of different polarity at room temperature. The bands at 320 and 400 nm are attributed to the enol and keto forms, respectively. | ||
In polar-protic solvents (methanol and ethanol) the keto tautomer (λmax = 400 nm) was considerably formed in compound 4, although the enol species (λmax = 320 nm) remained preferentially stabilized.21 Conversely, the intensity of the absorption bands indicated that the keto tautomer was slightly favored in methanol and ethanol for the derivative 5 even though a significant amount of the enol form was detected. In addition, in aprotic solvents as toluene and dimethylformamide, both compounds were predominantly stabilized in their enol form (λmax = 320 nm) with a band of much less intensity corresponding to the keto form (λmax = 400 nm) also observed.16b,c
The spectral data showed that the keto form is moderately enhanced by protic-polar solvents and much less favored in aprotic ones. This evidence suggests that the characterization of the Schiff bases 1–8 in CDCl3 solution (see ESI†) corresponds to an averaged structure (zwitterionic)7a from contributions of their enol and the keto species established in function of the nature of the solvent,14a–c,17 which seems a recurrent phenomenon in the solution NMR characterization of related compounds.7a
We investigated further the equilibrium in solution taking advantage of the structural simplicity of compounds 4–5, where a zwitterionic character is assumed in solution. It is well known that the 1H-chemical shift of the salicylidene OH signal of the Schiff bases is strongly dependent on their tautomeric structures18 with highly shifted signals being related to the keto character. In this series of compounds the salicylidene OH-chemical shift in CDCl3 was in the range from 13.18 to 16.75 ppm. Accordingly, 13C NMR spectra of all compounds showed the resonances corresponding to the salicylidene C2 atoms ranging from 160.5 to 173.7 ppm at room temperature. Therefore, we performed a solution VT NMR study of these compounds to investigate the effect of the solvent over the proton transfer process as a function of temperature10a,19 in the temperature range from 200 to 400 K.
With regard to compound 4 (Fig. 2a), the salicylidene OH signal reached the maximum chemical shift at the lowest temperature, indicating that the intramolecular hydrogen bond (O–H⋯N / N–H⋯O) is evidently strengthened.20 Conversely, this signal was shifted to lower frequencies at room temperature. These data reveals that the zwitterionic character of 4 is increasingly shifted toward its keto form as the temperature is decreased.10a,19 Evidently, the solvent also contributes to the stabilization of the keto forms,9c since the highest OH-chemical shift was observed in the solvent with the highest polarity (DMF-d7) as compared to CD2Cl2. This effect is based on the better stabilization of the keto form through electrostatic interactions (Scheme 1) as has been demonstrated by experimental and theoretical studies.10a,b,21
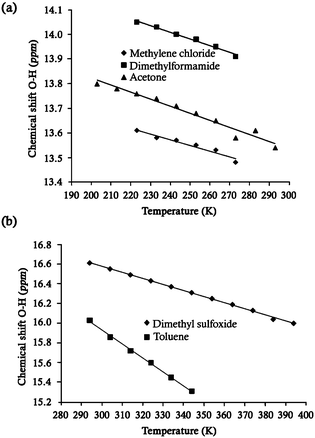
As we have previously pointed out, the salicylideneimine compounds in solution are generally best described as their averaged tautomeric forms at room temperature. However, the 1H and 13C spectra of compound 5 in CDCl3 at room temperature showed signals at 16.75 and 166.9 ppm for the salicylidene OH and C2 atoms, respectively, which are characteristic of derivatives with a marked keto character.14c,19c For this reason, compound 5 also was studied by VT NMR experiments using toluene-d8 and DMSO-d6. As expected, above room temperature the tautomeric equilibrium shifts towards the enol form. A shift of the salicylidene OH signal to low fequencies was observed as the temperature was increased (Fig. 2b), in accordance with the weakening effect of the temperature on the intramolecular hydrogen-bonding.22 Unfortunately, heating the imine 5 above 344 K in DMSO-d6 promotes a continuous hydrolysis that reaches a 15% ratio of hydrolyzed product at 394 K (see ESI†). The effect of the temperature in pyridine-d5 was also tested but the results showed no clear relation with the OH-chemical shift, due to the competition between the pyridine (solvent) and the imine nitrogen atom (compound 5) for the mobile proton.
Characterization in the solid-state of compounds 1–8
The IR analysis of crystalline compounds 1–8 obtained from methanol gave the first indication of the tautomeric form present. Characteristic bands were assigned in accordance with the literature data.23 Compounds 1–3 showed a sharp band around 3441–3316 cm−1 due to the ν(OH) vibration, whereas compounds 4–7 present a broad band assignable to the ν(NH) vibration around 3214–3053 cm−1. These data indicate the keto character of compounds 4–7 and the enol form in compounds 1–3. Bands for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds are in the range of 1627 to 1614 cm−1 in enol tautomers, while band for CO bonds in the keto species range from 1657 to 1639 cm−1.
N bonds are in the range of 1627 to 1614 cm−1 in enol tautomers, while band for CO bonds in the keto species range from 1657 to 1639 cm−1.
In the case of compound 8 the IR spectrum suggested the presence of both tautomeric species showing broad bands in all regions of the spectrum. Two bands above 3212 cm−1 may correspond to O–H⋯N, N–H⋯O and/or O–H⋯O interactions, and a very broad band at 1600 cm−1 can be attributed to the concomitant CO and CN bonds of keto and enol tautomers. X-Ray experiments confirmed the presence of both forms in the crystal of compound 8, as described in a further section.
X-Ray diffraction studies of compounds 1–7
After the initial experiments in solution, a detailed analysis of the molecular and packing motifs was carried out. Suitable crystals for X-ray diffraction24 of all compounds (1–8) were grown by slow evaporation of the methanolic solutions at room temperature. The most relevant crystallographic parameters are shown in Tables 1 and 2.| Crystal data a | 1 | 2 d | 3 | 4 |
|---|---|---|---|---|
| a λ Mo-Kα = 0.7103 Å. b R = ∑(Fo2 − Fc2)/∑Fo2. c R w = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. d The OH-benzyl and 5-tbutyl groups of molecule 2 are disordered over two positions, with SOFs for the major components of 76% and 52%, respectively. | ||||
| Formula | C22H35NO2 | C22H29NO2 | C21H19NO2 | C14H19NO2 |
| MW/g mol−1 | 345.51 | 339.46 | 317.37 | 233.30 |
| Crystal system | Monoclinic | Triclinic | Orthorhombic | Orthorhombic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P212121 | Pca21 |
| a/Å | 14.5103(2) | 10.2518(2) | 6.1478(2) | 9.0270(18) |
| b/Å | 28.8821(4) | 10.4845(2) | 7.5477(2) | 6.2300(12) |
| c/Å | 10.2616(1) | 19.6987(4) | 36.0798(1) | 22.198(4) |
| α (°) | 90 | 95.9376(7) | 90 | 90 |
| β (º) | 92.184(4) | 104.1553(8) | 90 | 90 |
| γ (°) | 90 | 92.2975(8) | 90 | 90 |
| V/Å3 | 4297.39(9) | 2037.31(7) | 1674.17(7) | 1248.4(4) |
| Z | 8 | 4 | 4 | 4 |
| ρ c/g cm−3 | 1.068 | 1.107 | 1.259 | 1.241 |
| Collected Refl. | 32630 | 23875 | 4795 | 4269 |
| Ind. Ref. (Rint) | 9747 (0.0601) | 9270 (0.0575) | 1958 (0.0476) | 1399 (0.0462) |
| Observed Ref. | 5426 | 5174 | 1312 | 1051 |
| R[I > 2σ(I)]b | 0.0634 | 0.0719 | 0.0499 | 0.0532 |
| R w (all data)c | 0.1910 | 0.2251 | 0.1488 | 0.1413 |
| Δρmax/e Å−3 | 0.25 | 0.28 | 0.20 | 0.28 |
| Δρmin/e Å−3 | −0.26 | −0.39 | −0.19 | −0.28 |
| Crystal data a | 5 d | 6 | 7 | 8 e |
|---|---|---|---|---|
| a λ Mo-Kα = 0.7103 Å. b R = ∑(Fo2 − Fc2)/∑Fo2. c R w = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. d The methyl group is equally disordered over two positions (SOF = 50%). e The diethylamino group of the molecule 8 is disordered over two positions, SOF = 57% for the major component. | ||||
| Formula | C10H13NO2 | C16H16N2O4 | C16H16BrNO2 | C18H21ClN2O2 |
| MW/g mol−1 | 179.21 | 300.31 | 334.21 | 332.82 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/c | P1 | P1 |
P
|
| a/Å | 10.0724(2) | 6.1083(2) | 7.2044(2) | 9.5103(2) |
| b/Å | 8.2670(17) | 10.3445(4) | 9.3402(2) | 12.8864(4) |
| c/Å | 11.7803(2) | 12.3439(5) | 12.3377(3) | 15.2106(4) |
| α (°) | 90 | 93.254(2) | 69.1986(10) | 97.8703(11) |
| β (°) | 111.75(3) | 100.417(2) | 80.3396(9) | 107.113(1) |
| γ (°) | 90 | 92.940(2) | 75.9081(9) | 100.4799(14) |
| V/Å3 | 911.0(3) | 764.38(5) | 749.70(3) | 1715.51(8) |
| Z | 4 | 2 | 2 | 4 |
| ρ c/g cm−3 | 1.307 | 1.305 | 1.480 | 1.289 |
| Collected Refl. | 3178 | 7278 | 12177 | 14135 |
| Ind. Ref. (Rint) | 1596 (0.0346) | 3435 (0.0517) | 5691 (0.0305) | 7334 (0.0393) |
| Observed Ref. | 1186 | 2447 | 4738 | 4472 |
| R[I > 2σ(I)]b | 0.0350 | 0.0613 | 0.0432 | 0.0858 |
| R w (all data)c | 0.1049 | 0.1787 | 0.1158 | 0.2786 |
| Δρmax/e Å−3 | 0.11 | 0.25 | 0.80 | 0.48 |
| Δρmin/e Å−3 | −0.17 | −0.23 | −0.48 | −0.49 |
Compounds 1 and 5 crystallize in the monoclinic space groupP21/c containing eight and four molecules per unit cell, respectively. Compounds 3 and 4 crystallize in the orthorhombic space groupsP212121 and Pca21, respectively, with four molecules per unit cell. Finally, compounds 2, 6 and 7 crystallize in the triclinic system, with four molecules per unit cell in the imine 2 (P), and two crystallographically independent molecules in the imines 6 and 7 (P1).
In all cases, the analysis of the Fourier maps confirmed the results suggested by the IR experiments. In compounds 1–3 the H1 atom was covalently bonded to the phenol moiety and in compounds 4–7 the analysis indicated the formation of keto tautomers, with the H1 atom completely transferred to the nitrogen atom forming an enamine group.
In aromatic Schiff bases, the C2–O1 and the C8–N9 bond distances are the main structural parameters to distinguish between tautomers. For enol tautomers 1–3, the C2–O1 bond distances were 1.349(2)/1.354(2) Å in 1a/1b, 1.354(2)/1.360(2) in 2a/2b, and 1.352(4) Å in 3. While the C8N9 bond distances are 1.268(2)/1.282(2) Å in 1a/1b, 1.281(2)/1.279(2) Å in 2a/2b and 1.276(4) Å in 3. All in agreement with the reported bond distances of related phenol tautomers25 and close to the averaged value for a C–O single bond (1.36 Å) and CN double bonds (1.28 Å).26 Conversely, the crystal structures of compounds 4–7 clearly showed a keto structure27 characterized by the shortening of the C2–O1 bond distances, 1.294(5) Å in 4, 1.314(2) Å in 5, 1.289(5)/1.268(5) Å in 6a/6b and 1.282(7)/1.286(7) Å in 7a/7b.
Moreover, the C–C bond lengths of the salicylidene ring (C2–C7) in enol tautomers 1–3 showed similar values, whereas in compounds 4–7, the bond distances reveal an alternating shortening and lengthening of the C–C bonds (Table 3). These results are in complete agreement with the assigned tautomeric forms.
| C2–C3 | C3–C4 | C4–C5 | C5–C6 | C6–C7 | C2–C7 | |
|---|---|---|---|---|---|---|
| a Two bond distances values are given when two crystallographically independent molecules are present in the asymmetric unit. Parameters correspond to (*) the enol tautomer and (+) the keto tautomer. | ||||||
| 1a* | 1.408(3) | 1.388(3) | 1.401(3) | 1.374(3) | 1.395(3) | 1.403(2) |
| 1b* | 1.409(3) | 1.390(2) | 1.393(3) | 1.375(3) | 1.396(2) | 1.401(3) |
| 2a* | 1.411(3) | 1.384(3) | 1.402(3) | 1.376(3) | 1.399(3) | 1.403(3) |
| 2b* | 1.404(3) | 1.383(3) | 1.402(3) | 1.369(3) | 1.398(3) | 1.400(3) |
| 3* | 1.386(5) | 1.377(5) | 1.396(5) | 1.362(5) | 1.396(5) | 1.408(5) |
| 4 + | 1.416(5) | 1.366(6) | 1.397(6) | 1.361(6) | 1.405(6) | 1.428(5) |
| 5 + | 1.415(2) | 1.368(2) | 1.390(2) | 1.367(2) | 1.412(2) | 1.429(2) |
| 6a + | 1.430(6) | 1.346(6) | 1.395(6) | 1.380(5) | 1.398(5) | 1.441(5) |
| 6b + | 1.437(6) | 1.358(6) | 1.407(5) | 1.366(5) | 1.401(5) | 1.443(5) |
| 7a + | 1.449(8) | 1.355(7) | 1.417(6) | 1.364(6) | 1.388(6) | 1.441(6) |
| 7b + | 1.443(7) | 1.376(7) | 1.410(7) | 1.369(6) | 1.390(7) | 1.427(7) |
Analysis of intramolecular interactions in compounds 1–7
The compounds herein reported showed two types of intramolecular hydrogen bonds, O1–H1⋯N9 in enol and N9–H9⋯O1 in the keto form. These interactions account for the formation of closely planar pseudoaromatic chelates S(6).28 Further examination of the O⋯N lengths in these intramolecular six-membered cycles for compounds 1–8 did not reveal a clear relationship with the tautomeric nature (A complete series of hydrogen-bonding parameters is listed in Table 4 of the ESI†). Regarding this, the well-known phenomenon called resonance-assisted hydrogen bonds (RAHBs)29 may explain the pronounced stability of the keto tautomers in terms of π-bond cooperativity effects of the conjugated system involved.30Analysis of the supramolecular arrangements for compounds 1–7
An exhaustive analysis of the structural parameters of compounds 1–7 indicated that the supramolecular structure in each tautomer is governed by one out of two intermolecular O–H⋯O hydrogen bonds. The O2–H2⋯O2 interaction, observed in the enol tautomers, associates closer molecules through aminoalcohol fragments (the aminoalcohol-aminoalcohol interaction), and the O2–H2⋯O1 interaction, present in keto tautomers, connects one aminoalcohol fragment with the ortho-hydroxy group of neighboring molecules through aminoalcohol-salicylidene interactions. Undoubtedly, the intermolecular hydrogen bonds contribute to the stabilization of the keto tautomers, which are less favored as isolated molecules.10a,31A detailed examination of the supramolecular arrangements in enol tautomers 1–2 showed that only the aminoalcohol-aminoalcohol interaction (O2–H2⋯O2) was involved. In compound 1, the expansion of this interaction generates infinite chains C(2) of molecules along the c crystallographic axis (Fig. 3a), with O⋯O distances of 3.003(2)/3.039(2) Å in 1a/1b molecules. In the case of compound 2, this interaction promoted by the benzylic hydroxy group, led to the formation of a tetrameric ring R44(8) (Fig. 3b), showing O⋯O distances of 2.49(3)/2.78(2) Å to 2a/2b molecules.
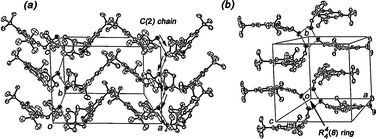 | ||
| Fig. 3 Crystal packing of enol tautomers 1 (a), and 2 (b). Ellipsoids are drawn at 50% probability. | ||
A shared characteristic of enol derivatives 1–2 is the 3,5-substitution by bulky tert-butyl groups that prevent the participation of the salicylidene fragment in intermolecular arrangements.
As a general trend, the ketonic salicylidene O1 atom commonly adopts the role of hydrogen-acceptor in the hydrogen-bonding geometry of the keto or zwitterionic tautomers, which is closely related to the high electrostatic charge of the heteroatoms (N and O)4,10a (Scheme 1).
Accordingly, in compounds 3–7, the only hydrogen-bonding interaction in their supramolecular structures is the aminoalcohol-salicylidene (O2–H2⋯O1) interaction. By expanding this interaction, infinite chains C(9) of molecules are generated along the a crystallographic axis in 3 (Fig. 4a) and along the b axis in 4 (Fig. 4b) and 5 (Fig. 4c). Additionally, an analogous interaction promotes the formation of a dimeric cycle R22(18) between two crystallographically independent molecules in imine 6 (Fig. 4d) and 7 (Fig. 4e). In compound 3, the participation of the enolic salicylidene O1 atom in the crystal packing was attributed to the acquired conformation in the fragment of the aminoalcohol by the presence of the bulky diphenyl unit.
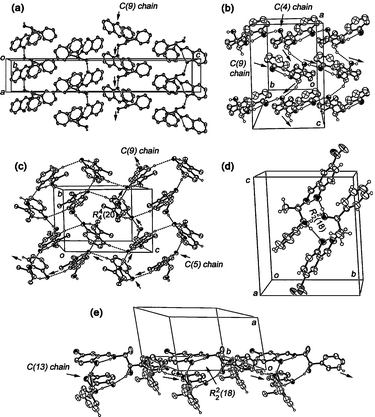 | ||
| Fig. 4 Crystal packing of enol tautomer 3 (a), keto tautomers 4 (b), 5 (c), 6 (d) and 7 (e). Ellipsoids are drawn at 50% probability. | ||
It is worth to notice that weaker C–H⋯O hydrogen bonds32 complement the packing of the tautomers (with exception of 3); however, the number in the case of the keto tautomers is larger than the one present in enol forms. In compound 4, the C10–H10B⋯O2 contacts form a C(4) chain of molecules that developed perpendicularly to the C(9) chain established by the O2–H2⋯O1 interactions (Fig. 4b). In compound 5, an expanded intramolecular ring R24(20) is formed by C6–H6⋯O1 interactions (Fig. 4c). Finally, intermolecular dimers were found in the packing of keto tautomer 7 linked through the C14B–H14B⋯O2A contacts, forming chains of dimers C(13)[R22(18)] along the c crystallographic axis (Fig. 4e).
In the case of the structurally related compounds 6–7, the electronwithdrawing nature of the para-substituted group (NO2 or Br) in the salicylidene fragment displaces the equilibrium exclusively to the keto form by completing proton transfer from the O1 to N9 atom. As expected, intermolecular interactions O2–H2⋯O1 complemented the stabilization of these keto tautomers. The O⋯O distances are 2.728(5)/2.725(4) Å in 6a/6b and 2.721(5)/2.719(5) Å in 7a/7b. Lastly, two additional Br⋯O contacts complete the sphere of coordination of the salicylidene O1 atom of compound 7 (ESI†).
X-Ray diffraction and solid-state NMR of compound 8
The X-ray diffraction analysis24 of compound 8 showed that the enol (labeled as 8A) and the keto (labeled as 8B) tautomers cocrystallized in the unit cell in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 5). The crystal structure has two intramolecular cycles S(6) that result from the O1A–H1A⋯N9A [dO1⋯N9 = 2.591(4) Å] and N9B–H9B⋯O1B [dO1⋯N9 = 2.623(3) Å] interactions. Moreover, the C2–O1 and C8–N9 bond distances attained values of 1.335(4) Å and 1.285(4) Å in the enol 8A and 1.303(3) Å and 1.328(3) Å in the keto form 8B, respectively. Similar bond distances were observed by Dong et al. in the cocrystallization of enol and keto tautomers from aza compounds.33
:1 ratio (Fig. 5). The crystal structure has two intramolecular cycles S(6) that result from the O1A–H1A⋯N9A [dO1⋯N9 = 2.591(4) Å] and N9B–H9B⋯O1B [dO1⋯N9 = 2.623(3) Å] interactions. Moreover, the C2–O1 and C8–N9 bond distances attained values of 1.335(4) Å and 1.285(4) Å in the enol 8A and 1.303(3) Å and 1.328(3) Å in the keto form 8B, respectively. Similar bond distances were observed by Dong et al. in the cocrystallization of enol and keto tautomers from aza compounds.33
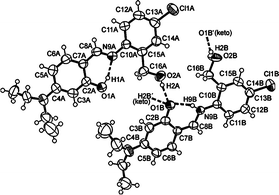 | ||
| Fig. 5 ORTEP drawing of the crystal structure of 8 showing one-half of the centrosymmetric arrangement where the enol and the keto tautomers co-crystallized in the unit cell. | ||
The crystal packing of derivative 8 (Fig. 5) is characterized by a centrosymmetric arrangement including the co-crystallization of both tautomers. The O(1B) atom of the keto species plays the role of triple hydrogen-bond acceptor and two adjacent OH-benzylic groups from enol-O(2A), keto-O(2B) and the N(9B) atoms are the corresponding donors. Two keto molecules form an expanded dimeric array (not shown) R22(20) by means of O2B–H2B⋯O1B′ interaction (dO2B⋯O1B = 2.702(4) Å). Furthermore, two neighboring OH-benzylic groups from enol molecules interact with keto-O(1B) atoms through O2A–H2A⋯O1B contacts (dO2A⋯O1B = 2.735(4) Å).
As an additional evidence supporting the coexistence of the two tautomeric forms in the crystal of 8, its 13C CPMAS spectrum was obtained from the same samples studied by X-ray crystallography. Solid-state 13C CPMAS NMR is a powerful technique capable of distinguishing between crystallographic entities such as polymorphs,34 solvates and hydrates,35 providing additional information, such as the number of molecules in the asymmetric unit.14,36
As noted in Fig. 6, a set of doublets of equal intensity for each expected signal confirmed the presence of two molecules per asymmetric unit. The 13C CPMAS dipolar dephasing experiment (to distinguish quaternary carbon atoms by suppressing any CH and CH2 signals) complemented the tentative assignment of the enol and keto tautomers signals. Two aromatic resonances at δ 177.7 and δ 163.6 that remained with equal intensity after the NQS experiment were assigned to CO and C–OH carbon atoms, respectively, in agreement with known shifts for the keto and enol forms.
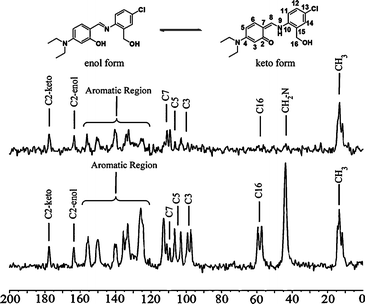 | ||
| Fig. 6 Below: solid-state 13C CPMAS spectra of compound 8. Above: 13C CPMAS dipolar dephasing experiment showing only quaternary and methyl carbons. | ||
An analogous analysis of the solid state spectra of compounds 5–6 (see ESI†) further support our carbonyl assignments. The signal at δ = 179.5 for compound 6 corresponds to the two crystallographically independent carbonyl groups found in the asymmetric unit. Additionally, a single peak assigned to CO at δ = 178.7 was observed in the spectrum of derivative 5 where one crystallographically independent keto molecule was present per asymmetric unit. It therefore became clear that the stabilization of the keto forms in this aggregate is enhanced by the multiple O2–H2⋯O1 hydrogen bonds, which are common interactions in the formation of the keto species.
Conclusions
The synthesis and the crystallographic study at room temperature of eight Schiff bases is reported. In the solid-state, each tautomer was characterized by different intermolecular O–H⋯O hydrogen bonds. The O2–H2⋯O2 interactions (aminoalcohol-aminoalcohol) were principally related to the enol forms in compounds 1 and 2, and the O2–H2⋯O1 interactions (aminoalcohol-salicylidene) were fundamentally associated with the keto species in compounds 4–7. Although, the enol tautomer was also observed in the case of derivative 3, voluminous substituents (i.e.diphenyl unit in the side of the aminoalcohol) prevent the formation of the expected O2–H2⋯O2 bonds and force the formation of O2–H2⋯O1 interactions. Additionally, the co-existence in the solid-state of both the keto and enol tautomers in 8 was supported by X-ray diffraction and solid-state NMR experiments. Solution UV-Vis experiments and solution VT NMR studies clearly revealed that the predominance for one tautomer is strongly dependent on the solvent investigated and the temperature used. Keto forms are preferred in protic solvents whereas enol tautomers were observed in aprotic ones. In spite of that, the preferential formation of a particular tautomer in the solid state is modulated through a combination of the steric factors of the substituents modifying the consequent capability of the molecules to interact with each other through appropriate intermolecular O–H⋯O hydrogen bonds.Experimental
Melting points were obtained with an Electrothermal 9200 apparatus and are uncorrected. Infrared spectra were measured on a FTIR Varian spectrophotometer ATR. 1H and 13C spectra as well as Correlation Spectroscopy 1H-1H COSY and Heteronuclear Chemical Shift Correlation 1H-13C HETCOR were recorded on JEOL eclipse +400 and ECA +500 spectrometers. Chemical shifts (ppm) are relative to (CH3)4Si for 1H and 13C. Mass spectra were recorded on an Agilent G1969A APCI Atmospheric Pressure-Chemical Ionization time-of-flight spectrometer. UV-Vis spectra were recorded on a Perkin Elmer Lambda 900 spectrophotometer.Solid-state NMR experiments
Crystalline samples of selected compounds obtained from methanolic solutions were previously ground with a mortar and a pestle at room temperature in order to get homogeneity, and they were packed in a 4 mm wide ZrO2 rotor with a KelF cap using 80–90 mg of each compound. 13C CPMAS solid state NMR spectra of selected compounds were obtained on a Bruker AVANCE II300 spectrometer operating at 13C frequency of 75.47 MHz with 1H broadband decoupler using a 4 mm broadband probe. A spinning frequency of 10 kHz at the magic angle was found to be successful for the removal of spinning sidebands using an optimized cross polarization contact time of 1 ms. Dipolar dephasing experiments (13C CPMAS non-quaternary suppression) were carried out with a delay of 25 μs before turning the 1H decoupler on.Single crystal X-ray structure determinations
All diffraction data were measured using an Enraf Nonius Kappa-CCD diffractometer with graphite-monochromated λMo-Kα = 0.71073 Å. Frames were collected at T = 293 K via ω/φ rotation. Direct methods SHELXS-86 and SIR-2004 were used for structure solution and SHELXL-97 program package for refinement and data output.24 C–H hydrogen atoms were placed in geometrically calculated positions using a riding model. O–H and N–H hydrogen atoms have been localized by difference Fourier maps and their bond distances and isotropic temperature factors have been refined freely (In compound 3, one restraint was necessary to fix a long O1–H1 bond distance [1.16(4) Å] with a DFIX 0.84 instruction). Three and four restraints were automatically inserted by SHELXL to fix the origin of the lattice in the refinement of compounds 6 and 7, respectively. Fifteen restraints were required to make the C–N and C–C bond distances of the diethylamino groups identical (one of them disordered over two positions) of the two crystallographically independent molecules of compound 8. As indicated in the literature,37 in compounds 3, 4 and 6 the Friedel opposites were merged before final refinement (MERG 4). The absolute configuration of 3 and 6 was chosen on the basis of the known configuration of the starting materials, while the configuration of 4 was arbitrarily selected. The Flack parameter for compound 7 refined satisfactorily to 0.028(9). Figures were created with ORTEP-3 for windows version 2.02. Verification of hydrogen-bonding interactions in the crystal lattice was carried out with PLATON program under the WINGX interface.Acknowledgements
The authors thank CONACyT (project No. 83519) and UNAM (PAPIIT IN-214010) for financial support and the scholarship to O. Domínguez and B. Rodríguez-Molina. Thanks are given to Consejo Superior de la Investigación Científica en España for the Cambridge Crystallographic Data Base license, to M. L. Rodríguez and V. González for NMR spectra, G. Cuéllar for MS, M. A. Leyva for X-ray diffraction and Dr E. García-Báez for valuable discussion.Notes and references
- (a) Y. Gat and M. Sheves, J. Am. Chem. Soc., 1993, 115, 3772–3773 CrossRef CAS; (b) S. Ren, R. Wang, K. Komatsu, P. Bonaz-Krause, Y. Zyrianov, C.-E. McKenna, C. Csipke, Z. A. Tokes and E. J. Lien, J. Med. Chem., 2002, 45, 410–419 CrossRef CAS.
- (a) G. K. Patra and I. Goldberg, Cryst. Growth Des., 2003, 3, 321–329 CrossRef CAS; (b) M. Sliwa, S. Létard, I. Malfant, M. Nierlich, P. G. Lacroix, T. Asahi, H. Masuhara, P. Yu and K. Nakatani, Chem. Mater., 2005, 17, 4727–4735 CrossRef CAS; (c) M. Sliwa, A. Spangenberg, I. Malfant, P. G. Lacroix, R. Métivier, R. B. Pansu and K. Nakatani, Chem. Mater., 2008, 20, 4062–4068 CrossRef CAS; (d) E.-Q. Gao, Y.-F. Yue, S.-Q. Bai, Z. He and C.-H. Yan, Cryst. Growth Des., 2005, 5, 1119–1124 CrossRef CAS.
- (a) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC; (b) M.-D. Zhou, J. Zhao, J. Li, S. Yue, C.-N. Bao, J. Mink, S. L. Zang and F. E. Kühn, Chem.–Eur. J., 2007, 13, 158–166 CrossRef; (c) R. M. Moreno, M. Rosol and A. Moyano, Tetrahedron: Asymmetry, 2006, 17, 1089–1103 CrossRef CAS.
- (a) V. Jeseentharani, J. Selvakumar, A. Dayalan, B. Varghese and K.-S. Nagaraja, J. Mol. Struct., 2010, 966, 122–128 CrossRef CAS; (b) K. Ogawa, J. Harada, I. Tamura and Y. Noda, Chem. Lett., 2000, 528–529 CrossRef CAS; (c) S. Bilge, Z. Kiliç, Z. Hayvali, T. Hökelek and S. Safran, J. Chem. Sci., 2009, 121, 989–1011 CrossRef CAS.
- (a) J. L. Scott and K. Tanaka, Cryst. Growth Des., 2005, 5, 1209–1213 CrossRef CAS; (b) J. Harada, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1999, 121, 5809–5810 CrossRef CAS; (c) E. Hadjoudis and I.-M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 RSC.
- (a) H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC; (b) F. M. Raymo and S. Giordano, Org. Lett., 2001, 3, 3475–3478 CrossRef CAS; (c) L. Giordano, T. M. Jovin, M. Irie and E. A. Jares-Erijman, J. Am. Chem. Soc., 2002, 124, 7481–7489 CrossRef CAS; (d) S. J. Lim, J. W. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS; (e) J. L. Maldonado, Y. Pónce-de-Léon, G. Ramos-Órtiz, M. Rodríguez, M. A. Meneses-Nava, O. Barbosa-García, R. Santillan and N. Farfán, J. Phys. D: Appl. Phys., 2009, 42, 075102 CrossRef; (f) H. Fukuda, K. Amimoto, H. Koyama and T. Kawato, Org. Biomol. Chem., 2003, 1, 1578–1583 RSC.
- (a) P.-E. Hansen, Z. Rozwadowski and T. Dziembowska, Curr. Org. Chem., 2009, 13, 194–215 CrossRef CAS; (b) R. M. Claramunt, C. López, M.-D. Santa María, D. Sanz and J. Elguero, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 169–206 CrossRef CAS; (c) Y.-M. Hijji, B. Barare, A.-P. Kennedy and R. Butcher, Sens. Actuators, B, 2009, 136, 297–302 CrossRef; (d) A. Perona, D. Sanz, R. M. Claramunt and J. Elguero, Molecules, 2006, 11, 453–463 Search PubMed; (e) J. H. Chong, M. Sauer, B. O. Patrick and M. J. MacLachlan, Org. Lett., 2003, 5, 3823–3826 CrossRef; (f) E. Ito, H. Oji, T. Araki, K. Oichi, H. Ishii, Y. Ouchi, T. Ohta, N. Kosugi, Y. Maruyama, T. Naito, T. Inabe and K. Seki, J. Am. Chem. Soc., 1997, 119, 6336–6344 CrossRef CAS.
- (a) A. Özek, Ç. Albayrak, M. Odabaoğlu and O. Büyükgüngör, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o177–180 CrossRef; (b) C. C. Ersanli, M. Odabaoğlu, Ç. Albayrak and A. Erdönmez, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o264–266 CrossRef.
- (a) C.-J. Chang, T.-L. Shieh and H. G. Floss, J. Med. Chem., 1977, 20, 176–178 CrossRef CAS; (b) W. Schilf, B. Kamieńsky and T. Dziembowska, J. Mol. Struct., 2002, 602–603, 41–47 CrossRef CAS; (c) S. Sharif, G. S. Denisov, M. D. Toney and H.-H. Limbach, J. Am. Chem. Soc., 2006, 128, 3375–3387 CrossRef CAS; (d) Z. Rozwadowski and T. Dziembowska, Magn. Reson. Chem., 1999, 37, 274–278 CrossRef CAS.
- (a) K. Ogawa and J. Harada, J. Mol. Struct., 2003, 647, 211–216 CrossRef CAS; (b) Filarowski, A. Kochel, M. Kluba and F. S. Kamounah, J. Phys. Org. Chem., 2008, 21, 939–944 CrossRef; (c) M. Sauer, C. Yeung, J. H. Chong, B. O. Patrick and M. J. MacLachlan, J. Org. Chem., 2006, 71, 775–788 CrossRef CAS.
- M. Rodríguez, R. Santillan, Y. López, N. Farfán, V. Barba, K. Nakatani, E. V. García-Baez and I. I. Padilla-Martínez, Supramol. Chem., 2007, 19, 641–653 CrossRef CAS.
- K. Ogawa, Y. Kasahara, Y. Ohtani and J. Harada, J. Am. Chem. Soc., 1998, 120, 7107–7108 CrossRef CAS.
- (a) K. Ogawa, J. Harada, T. Fujirawa and S. J. Yoshida, J. Phys. Chem. A, 2001, 105, 3425–3427 CrossRef CAS; (b) T. Fujiwara, J. Harada and K. Ogawa, J. Phys. Chem. A, 2009, 113, 1822–1826 CrossRef CAS.
- (a) L. Antonov, W. M. F. Fabian, D. Nedeltcheva and F. S. Kamounah, J. Chem. Soc., Perkin Trans. 2, 2000, 1173–1179 RSC; (b) M. Kluba, P. Lipkowski and A. Filarowski, Chem. Phys. Lett., 2008, 463, 426–430 CrossRef CAS; (c) T. Dziembowska, M. Szafran, A. Katrusiak and Z. Rozwadowski, J. Mol. Struct., 2009, 929, 32–42 CrossRef CAS.
- (a) A. Koll and P. Wolschann, Monatsh. Chem., 1996, 127, 475–486 CAS; (b) A. Filarowski, T. Glowiak and A. Koll, J. Mol. Struct., 1999, 484, 75–89 CrossRef CAS; (c) A. Filarowski, A. Koll and T. Glowiak, Monatsh. Chem., 1999, 130, 1097–1108 CrossRef CAS.
- (a) M. Miura, J. Harada and K. Ogawa, J. Phys. Chem. A, 2007, 111, 9854–9858 CrossRef CAS; (b) R. Herzfeld and P. Nagy, Curr. Org. Chem., 2001, 5, 373–394 CrossRef CAS; (c) S. H. Alarcon, A. C. Olivieri, R. M. Cravero, G. Labadie and M. J. Gonzáles-Sierra, J. Phys. Org. Chem., 1995, 8, 713–720 CrossRef CAS.
- (a) J. Jánsky and A. Koll, Struct. Chem., 2004, 15, 353–361 CrossRef; (b) W. M. F. Fabian, L. Antonov, D. Nedeltcheva, F. S. Kamounah and P. J. Taylor, J. Phys. Chem. A, 2004, 108, 7603–7612 CrossRef CAS.
- (a) A. Filarowski, A. Koll, M. Rospenk and I. Krol-Starzomska, J. Phys. Chem. A, 2005, 109, 4464–4473 CrossRef CAS; (b) N. Wachter-Jurcsak and C. A. Detmer, Org. Lett., 1999, 1, 795–798 CrossRef.
- (a) I. Król-Starzomska, A. Filarowski, M. Rospenk, A. Koll and S. Melikova, J. Phys. Chem. A., 2004, 108, 2131–2138 CrossRef CAS; (b) T. Dziembowska, Z. Rozwadowski, A. Filarowski and P. E. Hansen, Magn. Reson. Chem., 2001, 39, S67–S80 CrossRef CAS.
- (a) P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef; (b) P. Gilli, V. Bertolasi, L. Pretto, L. Antonov and G. Gilli, J. Am. Chem. Soc., 2005, 127, 4943–4953 CrossRef CAS.
- A. Filarowski and I. Majerz, J. Phys. Chem. A, 2008, 112, 3119–3126 CrossRef CAS.
- (a) N. S. Golubev, S. N. Smirnov, P. M. Tolstoy, S. Sharif, M. D. Toney, G. S. Denisov and H. H. Limbach, J. Mol. Struct., 2007, 844–845, 319–227 CrossRef CAS; (b) X. C. Tang, M. J. Pikal and L. S. Taylor, Pharm. Res., 2002, 19, 484–490 CrossRef CAS.
- (a) J. W. J. Ledbetter, J. Phys. Chem., 1977, 81, 54–59 CrossRef; (b) G. Wojciechowski, P. Przybylski, W. Schilf, B. Kamieński and B. Brzezinski, J. Mol. Struct., 2003, 649, 197–205 CrossRef CAS; (c) V. P. Rybalkin, A. D. Dubonosov, E. N. Shepelenko, L. L. Popova, M. I. Makarova, A. V. Tsukanov, B. A. Bren' and V. I. Minkin, Russ. J. Org. Chem., 2002, 38, 1326–1330 CrossRef CAS.
- Structure solution: (a) G. M. Sheldrick, SHELXS-86—Program for crystal structure solution, University of Göttingen, Germany, 1986 Search PubMed; (b) M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS; (c) Data Refinement: G. M. Sheldrick, SHELXL-97—Program for the refinement of crystal structure, University of Göttingen, Germany, 1997 Search PubMed; (d) ORTEP-Graphics: L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 Search PubMed; (e) PLATON-Hydrogen bonding interactions: A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 Search PubMed; (f) WINGX-interface: L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 Search PubMed.
- (a) V. T. Kasumov, H. Türkmen, İ. Uçar, A. Bulut and N. Yaylı, Spectrochim. Acta, Part A, 2008, 70, 60–68 CrossRef; (b) A. Takashiro and E. Yasuaki, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4315–4317 CrossRef.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Typical Interatomic Distances: Organic Compounds in International Tables of Crystallography, IUCR, 1992, vol. C, pp. 790–811 Search PubMed.
- (a) X.-X. Sun, S.-L. Ma, H. B. Huang and C.-M. Qi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o87–88 CrossRef; (b) B. Kaitner and G. Pavlović, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 2573–2575 CrossRef.
- For hydrogen-bond motifs notations see: J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 Search PubMed.
- (a) P. Gilli, V. Bertolasi, L. Pretto, V. Ferreti and G. Gilli, J. Am. Chem. Soc., 2004, 126, 3845–3855 CrossRef CAS; (b) V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1991, 113, 4917–4925 CrossRef CAS.
- V. Bertolasi, L. Pretto, G. Gilli and P. Gilli, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 850–863 CrossRef.
- (a) S. D. Chatziefthimiou, Y. G. Lazarou, E. Hadjoudis, T. Dziembowska and I. M. Mavridis, J. Phys. Chem. B, 2006, 110, 23701–23709 CrossRef CAS; (b) K. Pyta, P. Przybylski, W. Schilf, B. Kołodziej, A. Szady-Chełmieniecka, E. Grech and B. Brzezinski, J. Mol. Struct., 2010, 967, 140–166 CrossRef CAS.
- (a) T. Steiner, Crystallogr. Rev., 1996, 6, 1–57 CAS; (b) G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS; (c) P. A. Wood, F. H. Allen and E. Pidcock, CrystEngComm, 2009, 11, 1563–1571 RSC.
- Z.-H. Wu, J.-P. Ma, X.-W. Wu, R.-Q. Huang and Y.-B. Dong, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o128–130 CrossRef.
- (a) R. K. Harris, J. Pharm. Pharmacol., 2007, 59, 225–239 CrossRef CAS; (b) M. R. Caira, Top. Curr. Chem., 1998, 198, 163–208 CAS.
- (a) S. M. Reutzel-Edens, J. K. Bush, P. A. Magee, G. A. Stephenson and S. R. Byrn, Cryst. Growth Des., 2003, 3, 897–907 CrossRef CAS; (b) G. A. Stephenson, J. G. Stowell, P. H. Toma, R. R. Pfeiffer and S. R. Byrn, J. Pharm. Sci., 1997, 86, 1239–1244 CrossRef CAS.
- R. K. Harris, S. A. Joyce, C. J. Pickard, S. Cadars and L. Emsley, Phys. Chem. Chem. Phys., 2006, 8, 137–143 RSC.
- (a) H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CrossRef CAS; (b) A. Białosńka and Z. Ciunik, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o120–o122 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR characterization and experimental procedures for the preparation of all compounds, figure with high temperature VT NMR data of compound 5 in DMSO-d6, table with a complete list of intra- and intermolecular contacts, additional figure for the crystal packing of compound 7. CCDC reference numbers 712147 (1), 712151 (2), 712153 (3), 712149 (4) 712150 (5), 712154 (6), 712155 (7) and 712152 (8). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0nj00179a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |