Self-incorporation of a luminescent neutral iridium(III) complex in different mesoporous micelle-templated silicas
Daniela
Aiello
a,
Anna Maria
Talarico
b,
Francesca
Teocoli
b,
Elisabeta I.
Szerb
b,
Iolinda
Aiello
b,
Flaviano
Testa
a and
Mauro
Ghedini
*b
aCentro di Eccellenza CEMIF.CAL, CR INSTM, Unità INSTM della Calabria, Dipartimento di Ingegneria Chimica e dei Materiali, 87036 Arcavacata di Rende (CS), Italy
bCentro di Eccellenza CEMIF.CAL, LASCAMM – CR INSTM, Unità INSTM della Calabria and LiCryl, CNR-INFM, Dipartimento di Chimica, Università della Calabria, 87036 Arcavacata di Rende (CS), Italy. E-mail: m.ghedini@unical.it; Fax: +39 984 492066; Tel: +39 984 492062
First published on 19th October 2010
Abstract
The neutral luminescent tris-cyclometallated 2-phenylpyridine iridium(III) complex, (fac-Ir(ppy)3, [Ir]) following a self-assembling procedure, has been successfully located in the cavities of mesostructured silica materials through a surfactant-mediated process. Two different structure-directing agents, the cationic cetyltrimethyl ammonium bromide (CTAB) and the non-ionic poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123) were tested. The structural features induced by the metal complex incorporation can be explained by comparing the newly synthesized hybrid mesostructured materials with the corresponding undoped samples which were similarly prepared. X-Ray powder diffraction (XRD), nitrogen sorption, scanning electron microscopy (SEM), thermogravimetric analysis (TGA) and UV-Vis spectroscopy were used to characterize the investigated materials. Medium-sized spherical particles of 800 nm were obtained using CTAB as a structure-directing agent whereas larger monolithic aggregates with a minimum dimension of 5–8 μm were obtained using P123. The new hybrids showed the typical hexagonal symmetry of analogously prepared materials. Moreover high luminescence quantum yield values were obtained for both hybrids as a result of a very good dispersion of the chromophore in the mesostructured matrices, thereby avoiding dramatic self-quenching phenomena. The approach described in this paper provides a simple synthetic way to prepare new luminescent silica-based materials by the inclusion of neutral metal-containing luminophores into the pores of a mesoporous hosting skeleton.
Introduction
An important topic in photonic nanocomposite materials science is the design and synthesis of functional materials, which simultaneously allow satisfaction of the whole set of characteristics required to achieve specific optical properties and performances.Currently, ordered mesostructured materials, prepared by the co-condensation of an appropriate inorganic source and the surfactant self-assembling species doped with an optically active compound, have attracted a great deal of interest for potential optical applications such as, for example, high sensitive sensors,1–4 optical switches,5,6 solar cells,7 frequency doublers,8 and solid-state laser materials.9
Mesostructured materials with a geometrically regular inorganic skeleton have been shown to be appropriate scaffolds for the confinement of dyes.10–13 Since several dyes undergo luminescence quenching phenomena at relatively high concentrations, preventing advantageous applications, a possible way to overcome this drawback is given by the mesostructured materials, i.e., materials in which the organic template is not removed from the cavities resulting from the synthetic process adopted. These materials present well-defined organic–inorganic phase segregation at the nanometre scale which may effectively prevent aggregation and the related luminescence quenching phenomena. Moreover, it is worth recalling further excellent advantages, such as increased mechanical stability and shielding of the incorporated dye against chemical, thermal or photochemical degradation provided by the scaffold itself.
The chemistry of cyclometallated iridium(III) complexes is a current topic of investigations because they display unique photophysical properties such as good photo- and thermal stability, high phosphorescence quantum efficiencies, relatively short lifetimes and simple colour tuning through ligand structure and control, and a set of key features for molecular-based light emitting devices applications.14–18 However, it is also noteworthy that the actual performances of these molecular materials can be often severely limited because of the reduced emission efficiency in the solid state, owing to a concentration-driven quenching.19
Although different approaches, such as the dispersion of the emitters in a polymeric matrix or the introduction of sterically hindered substituents in the auxiliary ligands20,21 have been explored to limit the concentration quenching effect, at present none of them seem to solve this problem satisfactorily. In this context, mesostructured materials can be a useful tool for the dispersion of the emitting iridium(III) complexes thus obtaining better performing iridium(III)-based luminescent materials. In fact, a homoleptic tris-cyclometallated iridium(III) surfactant has been recently employed in the synthesis of an amphiphile/silica co-assembled nanocomposite which was successfully used as an active layer for organic light-emitting diode. This provided a better performing device with respect to the one based on the pristine solid.22
In this paper, selected as representative of neutral cyclometallated iridium(III) emitters, the synthesis, characterization and photophysical properties of new hybrid mesostructured silica materials incorporating the highly luminescent fac-isomer of the neutral homoleptic tris-cyclometallated 2-phenylpyridine iridium(III) complex,23fac-Ir(ppy)3 where Hppy = 2-phenylpyridine, which will then be labelled as [Ir] (Fig. 1), are reported.
![Chemical structure of [Ir].](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f1.gif) | ||
| Fig. 1 Chemical structure of [Ir]. | ||
The new materials described and discussed herein are obtained by a one-step procedure, similar to that developed by Zhou and Honma24, co-assembling the neutral luminescent [Ir] complex with a surfactant, which acts as the structure-directing agent, and tetraethoxysilane (TEOS) as an inorganic source. The selected surfactants are the cationic cetyltrimethyl ammonium bromide (CTAB) and the non-ionic triblock copolymer poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123). Moreover, because of the negligible solubility of [Ir] in H2O, EtOH has been used as co-solvent in order to increase, at least slightly, the [Ir] solubility and facilitate its inclusion within the hydrophobic regions of the surfactant during the template self-assembly process.
The silica-based luminescent materials prepared according to the above summarized procedure, with the general formula SiO2([Ir]·CTAB) and SiO2([Ir]·P123), were structurally and photophysically investigated. The differences induced by (i) the nature of the reacted surfactant with respect to the neutral character of [Ir], (ii) the resulting structural differences induced by the use of ethanol as co-solvent in the synthetic procedure and (iii) the resulting structural differences induced by the introduction of the [Ir] luminescent complex will be discussed.
Experimental
Materials
Tetraethyl orthosilicate (TEOS, 98%), ammonium hydroxide (NH4OH, 29 wt% NH3 in water), poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123), hydrochloric acid (HCl 37%), and absolute ethanol (EtOH, 99.98%) were used as supplied by Sigma-Aldrich. Cetyltrimethyl ammonium bromide (CTAB, 99.8 wt%) was purchased from Alfa Aesar. Water was produced from a Milli-Q system. fac-Ir(ppy)3, [Ir], was synthesized according to the procedure reported in the literature.25Methods used for characterization
Low angle X-ray diffraction (XRD) measurements were performed in the 2θ range of 1.0°–10.0° using a Philips 1730/10 diffractometer using Cu Kα radiation (λ = 1.5416 Å) at 40 kV and 200 mA and at-a-step width of 0.005°. Nitrogen sorption isotherms were measured at 77 K on a Micromeritics ASAP 2010 porosimeter. The materials were outgassed for 12 h under vacuum at 423 K before the measurements. Specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) method and pore sizes by the Barrett–Joyner–Halenda (BJH) method.26,27 Scanning electron microscopy images (SEM) were obtained with FEI FP 2353/0X. The thermogravimetric analysis (TGA) was carried out between 298 K and 1093 K under airflow and at a heating rate of 10 K min−1 in a Perkin Elmer thermobalance. Infrared spectra (FT-IR) were recorded on KBr pellets using a Perkin Elmer Spectrum One FT-IR spectrophotometer. Steady-state emission spectra were recorded on a Horiba Jobin Yvon Fluorolog 3 spectrofluorimeter, equipped with a Hamamatsu R-928 photomultiplier tube. The emission quantum yields (Φ) of the samples were obtained by means of a 102 mm diameter integrating sphere coated with Spectralon® and mounted in the optical path of the spectrofluorimeter using a 450 W Xenon lamp as an excitation source, coupled with a double-grating monochromator for selecting wavelengths. Φ is a ratio between “number of photons emitted” and the “number of photons absorbed”. The number of photons absorbed was calculated by monitoring the scattered excitation peak first with the blank sample in the sphere (La) and then with the sample in the sphere (Lc). The differences in peak intensity between these two measurements are caused by sample absorption and thus the number of absorbed photons can be calculated as the differences of the integrated curves (La − Lc). The number of photons emitted is calculated as the difference between the integrated luminescence of the sample (Ec) and that of the blank (Ea) and is therefore (Ec − Ea). As a result, Φ = (Ec − Ea)/(La − Lc). The experimental uncertainty on the Φ was 5%. In particular, mesostructured SiO2(CTAB) and SiO2(P123) were used as blank samples for the determination of Φ of SiO2([Ir]·CTAB) and SiO2([Ir]·P123), respectively. Time-resolved measurements were performed using the time-correlated single-photon counting (TCSPC) option on the Fluorolog 3. NanoLED at 370 nm, fwhm < 200 ps, was used to excite the sample. Excitation sources were mounted directly on the sample chamber at 90° to a single-grating emission monochromator (2.1 nm mm−1 dispersion; 1200 grooves mm−1) and collected with a TBX-04-D single-photon-counting detector. The photons collected at the detector were correlated by a time-to-amplitude converter (TAC) to the excitation pulse. Signals were collected using an IBH Data Station Hub photon counting module and data analysis was performed using the commercially available DAS6 software (HORIBA Jobin Yvon IBH). Goodness of fit was assessed by minimizing the reduced Chi squared function (χ2) and visual inspection of the weighted residuals. The average lifetimes were calculated as 〈τ〉 = ∑i(Aiτi)/∑iAi.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3 CTAB:3.8 NH4OH:153 H2O:17.1 EtOH. The template solution was prepared by dissolving 1.09 g (0.003 mol) of CTAB in 10 mL (0.171 mol) of EtOH under magnetic stirring. Then, 27.5 mL (1.53 mol) of H2O and 2.49 mL (0.038 mol) of NH4OH were added to this solution. After 5 min, 2.21 mL (0.01 mol) of TEOS was added under rapid stirring. The mixture was stirred for 3 days at room temperature, then filtered, washed with H2O and dried at 343 K overnight.
:0.0174 P123:6 HCl:167 H2O:17.1 EtOH. 1 g (1.74 × 10−5 mol) of P123 was dissolved in 10 mL (0.171 mol) of EtOH, under vigorous stirring at room temperature. Then, 30 mL (1.67 mol) of H2O and 1.83 mL (0.060 mol) of HCl were added to the solution. Finally, 2.21 mL (0.01 mol) of TEOS was added under stirring. The resulting mixture was aged at 373 K for 24 h, then filtered, washed with H2O and dried at 333 K for 12 h.
:0.3 CTAB:3.8 NH4OH:153 H2O:17.1 EtOH:0.0016 [Ir]. CTAB (1.09 g, 0.003 mol) was dissolved in 10 mL (0.171 mol) of EtOH; [Ir] (0.01 g, 1.6 × 10−5 mol) was added to the reaction mixture, which was stirred at room temperature for 5 h. The obtained yellow suspension was filtered to remove the solid (mixture of undissolved [Ir] and CTAB). Then 27.5 mL (1.53 mol) of H2O and 2.49 mL (0.038 mol) of NH4OH were added to the resulting solution. After 5 min, 2.21 mL (0.01 mol) of TEOS was added under vigorous stirring. The mixture was stirred for 3 days at room temperature, then filtered, washed with H2O and dried at 343 K overnight.
:0.0174 P123:6 HCl:167 H2O:17.1 EtOH:0.0016 [Ir]. An ethanolic solution (10 mL, 0.171 mol) of P123 (1 g, 1.74 × 10−5 mol) was obtained under vigorous stirring. Then, 0.01 g (1.6 × 10−5 mol) of [Ir] was added. The resulting mixture was stirred at room temperature for 5 h. Then the solid product (mixture of undissolved [Ir] and P123) was filtered and 30 mL (1.67 mol) of H2O and 1.83 mL (0.060 mol) of HCl were added to the solution. Finally, 2.21 mL (0.01 mol) of TEOS was added while stirring at room temperature. The mixture was aged at 373 K for 24 h, then filtered, washed with H2O and dried at 333 K for 12 h.
:0.3 CTAB:3.8 NH4OH:153 H2O:17.1 EtOH. The template solution was prepared by dissolving 1.09 g (0.003 mol) of CTAB in 10 mL (0.171 mol) of EtOH under magnetic stirring. Then, 27.5 mL (1.53 mol) of H2O and 2.49 mL (0.038 mol) of NH4OH were added to this solution. After 5 min, 2.21 mL (0.01 mol) of TEOS was added under rapid stirring. The mixture was stirred for 3 days at room temperature, then filtered, washed with H2O and dried at 343 K overnight.
:0.0174 P123:6 HCl:167 H2O:17.1 EtOH. 1 g (1.74 × 10−5 mol) of P123 was dissolved in 10 mL (0.171 mol) of EtOH, under vigorous stirring at room temperature. Then, 30 mL (1.67 mol) of H2O and 1.83 mL (0.060 mol) of HCl were added to the solution. Finally, 2.21 mL (0.01 mol) of TEOS was added under stirring. The resulting mixture was aged at 373 K for 24 h, then filtered, washed with H2O and dried at 333 K for 12 h.
:0.3 CTAB:3.8 NH4OH:153 H2O:17.1 EtOH:0.0016 [Ir]. CTAB (1.09 g, 0.003 mol) was dissolved in 10 mL (0.171 mol) of EtOH; [Ir] (0.01 g, 1.6 × 10−5 mol) was added to the reaction mixture, which was stirred at room temperature for 5 h. The obtained yellow suspension was filtered to remove the solid (mixture of undissolved [Ir] and CTAB). Then 27.5 mL (1.53 mol) of H2O and 2.49 mL (0.038 mol) of NH4OH were added to the resulting solution. After 5 min, 2.21 mL (0.01 mol) of TEOS was added under vigorous stirring. The mixture was stirred for 3 days at room temperature, then filtered, washed with H2O and dried at 343 K overnight.
:0.0174 P123:6 HCl:167 H2O:17.1 EtOH:0.0016 [Ir]. An ethanolic solution (10 mL, 0.171 mol) of P123 (1 g, 1.74 × 10−5 mol) was obtained under vigorous stirring. Then, 0.01 g (1.6 × 10−5 mol) of [Ir] was added. The resulting mixture was stirred at room temperature for 5 h. Then the solid product (mixture of undissolved [Ir] and P123) was filtered and 30 mL (1.67 mol) of H2O and 1.83 mL (0.060 mol) of HCl were added to the solution. Finally, 2.21 mL (0.01 mol) of TEOS was added while stirring at room temperature. The mixture was aged at 373 K for 24 h, then filtered, washed with H2O and dried at 333 K for 12 h.
For the other mesostructured materials, SiO2(P123) and SiO2([Ir]·P123), the removal of the neutral surfactant P123 was carried out via chemical extraction using only EtOH (300 mL), which was added to 1 g of solid sample. The resulting mixture was stirred for 6 h at reflux. The obtained solids, abbreviated in a similar way to the other extracted materials, SiO2(P123)* and SiO2([Ir]·P123)*, respectively, were dried at 343 K overnight.
Results and discussion
Synthesis
The new mesostructured hybrid materials containing the luminescent neutral iridium(III) chromophore, SiO2([Ir]·CTAB) and SiO2([Ir]·P123) were synthesized using a one-pot procedure, via a modified synthetic strategy. In particular, the chromophore [Ir] was incorporated into the micelles formed by two different types of surfactants (cationic CTAB and non-ionic P123), using a H2O/EtOH mix solvent system, which furthermore acts as structure-directing agents for the synthesis of the final hybrid materials (Scheme 1). In order to investigate the differences induced by the use of ethanol as co-solvent, their analogous blank materials without the luminescent chromophore, SiO2(CTAB) and SiO2(P123) were also prepared, characterised and discussed with reference to those reported in the literature.28,29![A possible schematic illustration of [Ir] incorporation into the mesostructured silica materials.](/image/article/2011/NJ/c0nj00533a/c0nj00533a-s1.gif) | ||
| Scheme 1 A possible schematic illustration of [Ir] incorporation into the mesostructured silica materials. | ||
Furthermore, to investigate the surface properties after the inclusion of the luminescent iridium(III) chromophore, the porosity producing templates were completely removed (FT-IR evidences) by extraction with specific solvents. Comparison of the two types of extracted solids, blanks and hybrids was useful for understanding the surface effects in terms of diameters and pore volumes.
X-Ray diffraction investigations (XRD)
The XRD patterns of all the synthesized mesostructured materials (hybrids and blanks) were recorded (Fig. 2). Both SiO2(CTAB) and SiO2([Ir]·CTAB) exhibit XRD patterns similar to the ones reported in the literature for analogous systems.28 In particular, the patterns consist of small angle of three relatively broad reflections indexed as [100], [110] and [200], characteristic of a hexagonal lattice.![XRD patterns of (a) SiO2(CTAB) top and SiO2([Ir]·CTAB) bottom; (b) top—SiO2(P123) and bottom—SiO2([Ir]·P123).](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f2.gif) | ||
| Fig. 2 XRD patterns of (a) SiO2(CTAB) top and SiO2([Ir]·CTAB) bottom; (b) top—SiO2(P123) and bottom—SiO2([Ir]·P123). | ||
Similar interplanar distances d100, and consequently the similar hexagonal lattice constant a (Fig. 2a) indicate that the presence of [Ir] did not affect the self-assembly process during mesophase formation, leaving the mesostructure substantially unchanged. This phenomenon can be explained by the fact that the chromophore was introduced between the hydrophobic tails of the CTAB surfactant in a periodical manner within a mesostructured channel.30 However, the decrease in the intensity of the diffraction peaks, in the case of the hybrid material SiO2([Ir]·CTAB), may be indicative of a more disorganized material compared with the blank one. In addition both mesostructured materials containing CTAB showed a shrinkage of the d spacings typically observed when, instead of pure H2O, a H2O/EtOH mixture is employed in the synthesis.28
The XRD patterns of the SiO2(P123) and SiO2([Ir]·P123) samples (Fig. 2b) consisted of four peaks that can be indexed as [100], [110], [200] and [210] associated with a hexagonal symmetry. These patterns are similar to the ones exhibited by the analogues P123-template materials reported in the literature,29 with an expected shrinkage of the cell parameters because of the use of EtOH as co-solvent.28 Nevertheless the SiO2([Ir]·P123) sample shows an increase in the interplanar distance and hexagonal unit cell length with respect to the SiO2(P123) blank (Fig. 2b). This may be indicative of the placement of the [Ir] complex within the interior of the micellar aggregates, which consequently expands the micelles size.30 Furthermore, contrary to systems containing the CTAB surfactant, the inclusion of the iridium(III) complex seems to enhance slightly the order parameter of the hybrid material SiO2([Ir]·P123), as the increase in the intensity of the reflection peaks indicates when the patterns are compared with the blank SiO2(P123).
No significant changes were observed for the XRD patterns recorded several weeks later for both CTAB and P123-template materials stored in air, indicating that these structures were very stable.
Nitrogen sorption
The specific surface areas (SBET), pore diameters (dp) and pore volume (Vp) of all prepared materials were calculated by the BET method and BJH model, respectively (Table 1).The adsorption/desorption isotherms of SiO2(CTAB)*, SiO2([Ir]·CTAB)*, SiO2(P123)* and SiO2([Ir]·P123)* are shown in Fig. 3 and Fig. 4.
![Nitrogen adsorption and desorption isotherms for (a) SiO2(CTAB)* and (b) SiO2([Ir]·CTAB)*.](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f3.gif) | ||
| Fig. 3 Nitrogen adsorption and desorption isotherms for (a) SiO2(CTAB)* and (b) SiO2([Ir]·CTAB)*. | ||
![Nitrogen adsorption and desorption isotherms for (c) SiO2(P123)* and (d) (SiO2 ([Ir]·P123)*.](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f4.gif) | ||
| Fig. 4 Nitrogen adsorption and desorption isotherms for (c) SiO2(P123)* and (d) (SiO2 ([Ir]·P123)*. | ||
The isotherms of all the extracted samples are type IV according to the IUPAC classification, and typical of mesoporous materials with uniform size distribution. The porosimetric values dp, SBET, and Vp determined for all the extracted samples are coherent with that obtained for analogously synthesized materials.28,29 Nevertheless the values measured for SiO2([Ir]·CTAB)* and SiO2([Ir]·P123)* samples, including dp and Vp, are slightly increased with respect to the corresponding blank ones. Therefore, it is reasonable to assume that the self-inclusion of [Ir] inside the micelles during the co-assembly process affects their dimensions, thus leading to an enlargement of the resulting pore size and pore volume.
Scanning electron microscopy (SEM)
Fig. 5 shows representative SEM micrographs of the [Ir] solid. In the solid state this luminescent complex forms long fibres with a hexagonal section and medium diameter of 2–5 μm.![Representative SEM image of [Ir].](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f5.gif)
Fig. 6a–d shows SEM micrographs of all mesostructurated materials synthetised. The solids exhibit very different morphologies. Syntheses using H2O and EtOH as co-solvent permit spherical particles with ordered mesopores to be obtained. In fact, SiO2(CTAB) as SiO2([Ir]·CTAB) (Fig. 6a and b) consists of medium-sized spherical particles of 800 nm and a homogeneous size distribution. SiO2(P123) as SiO2([Ir]·P123) (Fig. 6c and d) samples exhibits larger and much more monolithic aggregates with a minimum dimension of 5–8 μm.
![Representative SEM image of SiO2(CTAB) (a), SiO2([Ir]·CTAB) (b), SiO2(P123) (c) and SiO2([Ir]·P123) (d).](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f6.gif) | ||
| Fig. 6 Representative SEM image of SiO2(CTAB) (a), SiO2([Ir]·CTAB) (b), SiO2(P123) (c) and SiO2([Ir]·P123) (d). | ||
Thermogravimetric analysis (TGA)
The thermal decomposition behaviour of all mesostructurated solids (hybrids and blanks) was studied using TGA measurements.In the range of 413–873 K, SiO2(CTAB) and SiO2(P123) samples exhibit a weight loss attributed to a surfactant decomposition of 44.8% and 36.5% respectively. In the same temperature range, SiO2([Ir]·CTAB) and SiO2([Ir]·P123) samples exhibit a weight loss of about 48.3% and 39.7%, which probably corresponds to the decomposition of [Ir] together with the surfactant. The similar weight losses indicate similar behaviours despite the different templates used in the synthesis. Nevertheless, the difference of weight loss resulting from a comparison among the blank and the corresponding hybrid materials (approximately 3%) cannot be attributed only to the presence of the [Ir] chromophore, and it probably arises from both [Ir] decomposition and an intrinsic difference of the weight ratio between the surfactants and the inorganic scaffold due to a different degree of order induced by the [Ir] co-assembling process. These results are supported also by XRD measurements and nitrogen sorption analysis, indicating that the presence of [Ir] in the micelles causes some variations in surface parameters.
Photoluminescence properties
It is well known that for iridium(III) complexes, luminescence quantum yield in the solid state is a function of aggregation. Aggregate formation often quenches light emission limiting the development of photoluminescent materials that exhibit strong solid-state emission properties.31 Nevertheless, in some cases, it has been demonstrated that the luminescence quantum yield of iridium(III) complexes in solid states, such as doped polymer films or powder states with suitable supramolecular organization of the chromophores, is higher than that in solutions.32 Specifically in the case of [Ir], it has been shown that its photoluminescence efficiency can vary from 0.0098 in neat film, to 0.7 in solution, reaching a value of 0.9 in highly dispersed polymeric matrix.33In order to investigate the photoluminescence properties of the newly synthesized mesostructured materials, a full photophysical investigation including emission spectra, phosphorescence quantum yield and time-resolved luminescence was performed for comparison of the two hybrid solids and the pristine [Ir] powder. The full photophysical results compared to the photophysical properties of [Ir] complex in solution33,34 are summarized in Table 2.
| Materials | λ max/nm | Φ | τ Ph/(Ai) ns | τ rad/ns | 〈τ〉/ns | ϕ calc. |
|---|---|---|---|---|---|---|
| a Data concerning [Ir] in deoxygenated toluene solution taken from ref. 33 and 34. b Data concerning [Ir] in polystyrene matrix (1 wt%) taken from ref. 33. c [Ir] pristine powder. | ||||||
| [Ir] | 515 | 0.73 | 1090 | 1500 | — | — |
| [Ir] | — | 0.91 | 1200 | 1300 | — | — |
| [Ir] | 534 | 0.012 | τ 1 = 0.1 (49%) | — | 19 | ∼0.014 |
| τ 2 = 38 (50%) | ||||||
| SiO2([Ir]·CTAB) | 509 | 0.3 | τ 1 = 576 (27%) | — | 931 | ∼0.72 |
| τ 2 = 1077 (72%) | ||||||
| SiO2([Ir]·P123) | 515 | 0.04 | τ 1 = 527 (25%) | — | 983 | ∼0.75 |
| τ 2 = 1120 (76%) |
In Fig. 7 are reported the emission spectra of the mesostructured powders SiO2([Ir]·CTAB) and SiO2([Ir]·P123) compared to the emission spectra of the [Ir] powder. With reference to the emission spectra of both SiO2([Ir]·CTAB) and SiO2([Ir]·P123) no red shift of the emission maxima is observed on moving from solution to the two different mesostructured powders whereas a substantial red shift is observed for pristine [Ir] solid.
![Emission spectra of SiO2([Ir]·CTAB) (dash line), SiO2([Ir]·P123) (dotted line) and of [Ir] pristine powder (solid line).](/image/article/2011/NJ/c0nj00533a/c0nj00533a-f7.gif) | ||
| Fig. 7 Emission spectra of SiO2([Ir]·CTAB) (dash line), SiO2([Ir]·P123) (dotted line) and of [Ir] pristine powder (solid line). | ||
Since a significant red shift of the emission maxima is expected for aggregation states which favour the occurrence of a high degree of interchromophore contacts,35 the observed spectral features seem to indicate (i) a substantial absence of strictly interacting iridium(III) moieties in the mesoporous powders as a result of a very good dispersion of the host in the matrices and (ii) the existence of a high degree of interchromophore contact in the pristine solid.
Regarding phosphorescence quantum efficiency, the [Ir] solid shows considerably reduced phosphorescence quantum yield with respect both to solutions and dispersed polymeric matrix (Table 2). Moreover, its luminescence decay turned out to be considerably fast and non-exponential thus indicating the presence of additional non-radiative decay channels caused by aggregation in the solid state.
Differently from the [Ir] solid, SiO2([Ir]·CTAB) and SiO2([Ir]·P123) showed much longer luminescence lifetimes, even if still non-exponential. In particular both solids showed a longer lifetime component of ∼1100 ns, which account for most of the signal (75%) and a shorter one of ∼500 ns accounting for 25% of the total decay.
The longer luminescent lifetime component is comparable with that observed for [Ir] highly dispersed in a polymeric rigid matrix (Table 2) which is usually associated with the lifetime of the isolated rigid molecule. Therefore this component could be attributed to a large fraction of non-interacting iridium(III) moieties reasonably included in the interior of the micelle cores where the chromophore is preferentially shielded from aggregation and consequently from the luminescence quenching phenomena. Furthermore, the faster lifetime component may result from a fraction of iridium(III) chromophore whose luminescence can be quenched through the exciton migration quenching phenomena between closely packed iridium(III) chromophores and/or iridium chromophores and impurity traps possibly present in the materials.
Phosphorescence quantum yields of SiO2([Ir]·CTAB) and SiO2([Ir]·P123) mesostructured solids were determined following the procedure described in the Experimental section. Different results were obtained for the two solids (0.3 and 0.04 respectively) despite the very similar photophysical data derived from both steady state and time-resolved luminescence measurements. Moreover, these values disagree significantly with the value reported for the [Ir] complex dispersed in polystyrene matrix (Table 2) despite the similarity of lifetimes values. Therefore, our measured phosphorescence quantum yields should be affected by instrumental artefacts likely to arise from important light scattering phenomena typical of such materials and affecting substantially the calculated values of absorbed photons. Nevertheless, the phosphorescence quantum yields (ϕp) were roughly estimated. When only one type of emitting species is present in the sample, the phosphorescence lifetime (τp) and ϕp values are related to the radiative lifetime (τr) of the emissive state as:
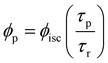
Considering the value reported in the literature for [Ir] dispersed in a polystyrene matrix as the radiative lifetime and the medium values 〈τ〉 calculated from experimental data as the phosphorescence lifetime, the phosphorescence quantum yields obtained for SiO2([Ir]·CTAB) and SiO2([Ir]·P123) are 0.72 and 0.75, respectively. Moreover, the phosphorescence quantum yield of the [Ir] pristine solid was calculated analogously obtaining a result which was comparable to that measured (Table 2).
The calculated luminescence quantum yield values suggest that the experimental approach (technique, set-up) adopted for their determination suffers from a severe limitation arising from the non-negligible scattering phenomena displayed by the investigated materials. However, the calculated values seem to agree with both XRD and porosimetric results, showing the [Ir] complexes to be mainly included within the interior of the micellar aggregates.
Conclusions
These investigations were made in order to test a synthesis protocol which can be beneficial for the preparation of new emitting materials for optical applications. In particular, in order to evaluate the possible effects induced by the different electrostatic interactions that could take place between the templating agent and the neutral [Ir] compound, mesostructured materials containing a cationic or a non-ionic surfactant have been prepared. The adopted approach allowed for the incorporation of the [Ir] guest into a mesostructured matrix host by a co-organizing process involving the two selected surfactants: the cationic CTAB and the non-ionic P123. The XRD and nitrogen sorption measurements of the obtained materials evidenced the preferential inclusion of the iridium(III) chromophore into the hydrophobic channels of the mesostructure without any substantial changes to the typical hexagonal symmetry, with respect to both the blank samples and analogous synthesized materials. Nevertheless, XRD measurements showed that when the cationic CTAB is used as a structure directing agent, the [Ir] complex is preferentially placed in between the hydrophobic tails of the surfactants. In contrast, the non-ionic P123 directs the chromophore mainly into the centre of micelle cores.Moreover, the resultant hybrid materials exhibit the characteristic emission of the [Ir] complex with a high luminescence quantum yield originating from the effective good dispersion of the chromophore within the mesostructured matrices. Further investigations are currently under way in order to extend the described approach to ionic chromophores.
Acknowledgements
The work was supported by the Consorzio Interuniversitario Nazionale per la Scienza e la Tecnologia dei Materiali (INSTM) and the Ministero dell'Istruzione, dell'Università e della Ricerca (MiUR), through the PRISMA 2007 (PC26/2007) and the Progetti di Ricerca di Interesse Nazionale (PRIN 2007-2007WJMF2W).References
- T. A. Dickinson, J. White, J. S. Kauer and D. R. Walt, Nature, 1996, 382, 697 CrossRef CAS.
- H. Y. Fan, Y. F. Lu, A. Stump, S. T. Reed, T. Baer, R. Schunk, V. Perez-Luna, G. P. Lopez and C. J. Brinker, Nature, 2000, 405, 56 CrossRef.
- G. Wirnsberger, B. J. Scott and G. D. Stucky, Chem. Commun., 2001, 119 RSC.
- B. H. Han, I. Manners and M. A. Winnik, Chem. Mater., 2005, 17, 3160 CrossRef CAS.
- B. Schaudel, C. Guermeur, C. Sanchez, K. Nakatani and J. A. Delaire, J. Mater. Chem., 1997, 7, 61 RSC.
- M. Levitus and P. F. Aramendia, J. Phys. Chem. B, 1999, 103, 1864 CrossRef CAS.
- B. V. Bergeron, C. A. Kelly and G. J. Meyer, Langmuir, 2003, 19, 8389 CrossRef CAS.
- G. Ihlein, B. Junges, U. Junges, F. Laeri, F. Schüth and U. Vietze, Appl. Organomet. Chem., 1998, 12, 305 CrossRef CAS.
- G. Wirnsberger, P. D. Yang, B. J. Scott, B. F. Chmelka and G. D. Stucky, Spectrochim. Acta, Part A, 2001, 57, 2049 CrossRef CAS.
- P. Yang, G. Wirnsberger, H. C. Huang, S. R. Cordero, M. D. Mc Gehee, B. Scott, T. Deng, G. M. Whitesides, B. F. Chmelka, S. K. Buratto and G. D. Stucky, Science, 2000, 287, 465 CrossRef CAS.
- B. J. Scott, G. Wirnsberger and G. D. Stucky, Chem. Mater., 2001, 13, 3140 CrossRef CAS.
- E. Dovgolevsky, S. Kirmayer, E. Lakin, Y. Yang, C. J. Brinker and G. L. Frey, J. Mater. Chem., 2008, 18, 423 RSC.
- N. Mizoshita, Y. Goto, T. Tani and S. Inagaki, Adv. Mater., 2009, 21, 4798 CrossRef CAS.
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CAS.
- M. K. Nazeeruddin and M. Grätzel, Struct. Bonding, 2007, 123, 113 CAS.
- Y. You and S. Young Park, Dalton Trans., 2009, 1267 RSC.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS.
- Y. Kawamura, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Phys. Rev. Lett., 2006, 96, 017404 CrossRef.
- C. Rothe, C.-J. Chiang, V. Jankus, K. Abdullah, X. Zeng, R. Jitchati, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 2038 CrossRef CAS , and references therein.
- C.-H. Chang, C.-C. Chen, C.-C. Wua, C.-H. Yang and Y. Chi, Org. Electron., 2009, 10, 1364 CrossRef CAS.
- C.-H. Yang, S.-H. Yang and C.-S. Hsu, Nanotechnology, 2009, 20, 315601 CrossRef , and references therein.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
- H. S. Zhou and I. Honma, Adv. Mater., 1999, 11, 683 CrossRef CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373 CrossRef.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Chem. Mater., 2002, 14, 4721 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- H. S. Zhou, H. Sasabe and I. Honma, J. Mater. Chem., 1998, 8, 515 RSC , and references therein.
- Y. Kawamura, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Phys. Rev. Lett., 2006, 96, 017404 CrossRef.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267 RSC.
- W. Holzer, A. Penzkofer and T. Tsuboi, Chem. Phys., 2005, 308, 93 CrossRef CAS.
- E. B. Namdas, A. Ruseckas, I. D. W. Samuel, S. C. Lo and P. L. Burn, J. Phys. Chem. B, 2004, 108, 1570 CrossRef CAS.
- H. Wang, Q. Liao, H. Fu, Y. Zeng, Z. Jiang, J. Maa and J. Yao, J. Mater. Chem., 2009, 19, 89 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |