The cocrystal of 4-oxopimelic acid and 4,4′-bipyridine: polymorphism and solid-state transformations†
Ivan
Halasz
*,
Mirta
Rubčić
,
Krunoslav
Užarević
,
Ivica
Đilović
and
Ernest
Meštrović
Department of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: ihalasz@chem.pmf.hr
First published on 7th October 2010
Abstract
The cocrystal of 4-oxopimelic acid and 4,4′-bipyridine was isolated in two polymorphic forms depending upon the solvent of crystallisation. Polymorphs exhibit hydrogen-bonded chains formed between the carboxyl and pyridine moieties while different packing arrangements of chains result from C–H⋯O interactions. Solid-state synthesis and interconversion conditions were investigated by grinding and thermal methods.
Multicomponent molecular crystals, usually referred to as cocrystals,1 have recently come into focus due to their potential applications in e.g.pharmaceutical industry2 and synthetic chemistry.3 Their design relies on the predictability of supramolecular interactions between the complementary functionalities of cocrystal components. Among other methods, hydrogen bonding, with its established value in some homo- and heterosynthons, is often the tool of choice.4 This allows a significant degree of control in the construction of multicomponent organic materials and facilitates their primary structure prediction.5 However, even if the main supramolecular synthon can be predicted with a fair degree of certainty,6 this often provides no information about the influence of weaker interactions on the conformation and spatial arrangement of the cocrystal components.7 In this respect, investigation of the polymorphic cocrystals8 is of great importance, since, through conformational and structural differences, it allows insights into the role of weak intermolecular contacts in the packing of molecular assemblies.
In this context, cocrystals of pimelic acid (pa) and 4,4′-bipyridine (bipy), which have been previously found to exist in three polymorphic forms, represent an interesting example. Each of the three forms is characterized by one-dimensional chains held together by O–H⋯N hydrogen-bonds involving the pyridine nitrogen and the carboxylic group.9 Herein we report two conformational polymorphs of cocrystals of 4-oxopimelic acid (opa) and 4,4′-bipyridine (bipy) isolated by solvent crystallisation and their interconversions in the solid state.‡ This work was focused on exploring the influence of a competing hydrogen bond acceptor (keto group of opa) on the conformation and structure of the resulting self-assembled solids. To improve polymorph resolution,10 solid-state syntheses viagrinding and liquid-assisted grinding (LAG)11 were investigated. Interconversion conditions by thermal treatment and grinding were also examined.
In this system, the formation of 3 types of supramolecular synthons can be expected (Scheme 1). A CSD search12 revealed 795 entries containing at least one pyridine and one carboxyl moiety. Carboxyl-pyridine heterosynthon A occurs in 508 (64%) of the 795 structures, while only 37 (5%) structures exhibit exclusively carboxyl–carboxyl supramolecular homosynthon B. Exclusive appearance of heterosynthon C was found in a subset of 40 (5%) compounds (mostly the carboxyl-amide synthon). In a repeated search excluding potentially bulky groups on the pyridine moiety,13 the occurrence of the synthon A rose to 88% whereas the occurrence of synthons B and C was negligible. Therefore, the carboxyl-pyridine heterosynthon is the most probable supramolecular synthon to occur in cocrystal formation between opa and bipy (Scheme 1). Our analysis follows the trends found in previous studies of occurrence of supramolecular heterosynthons encompassing carboxylic and pyridine moieties.14
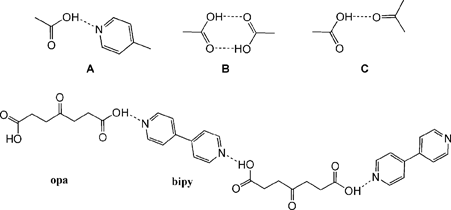 | ||
| Scheme 1 | ||
Two morphologically diverse colourless species of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 opa·bipy cocrystal were collected depending on the choice of the solvent. Form I was isolated as prisms by crystallisation from ethanol, while plates of Form II were obtained from methanol.‡ Further polymorph screening by solvent crystallisation revealed no other crystalline species (ESI†). The main supramolecular synthon governing cocrystal formation in both polymorphs is, as expected, the carboxyl-pyridine heterosynthon (Scheme 1A, Table 1), which leads to the formation of infinite one-dimensional (1-D) hydrogen-bonded chains which assume a zig-zag architecture (Fig. 1). To test if the heterosynthon A remains the dominant interaction when higher concentrations of the competing carbonyl group are present, a series of reactions were conducted whereby the excess of opa was varied up to 6:1. However, the sole product isolated from crystalization experiments was the 1:1 cocrystal (ESI†).
:1 opa·bipy cocrystal were collected depending on the choice of the solvent. Form I was isolated as prisms by crystallisation from ethanol, while plates of Form II were obtained from methanol.‡ Further polymorph screening by solvent crystallisation revealed no other crystalline species (ESI†). The main supramolecular synthon governing cocrystal formation in both polymorphs is, as expected, the carboxyl-pyridine heterosynthon (Scheme 1A, Table 1), which leads to the formation of infinite one-dimensional (1-D) hydrogen-bonded chains which assume a zig-zag architecture (Fig. 1). To test if the heterosynthon A remains the dominant interaction when higher concentrations of the competing carbonyl group are present, a series of reactions were conducted whereby the excess of opa was varied up to 6:1. However, the sole product isolated from crystalization experiments was the 1:1 cocrystal (ESI†).
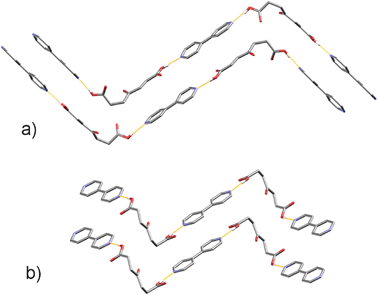 | ||
| Fig. 1 Hydrogen-bonded zig-zag chains in (a) Form I, and (b) Form II. Colour code: grey – carbon, red – oxygen, white – hydrogen, blue – nitrogen. Hydrogen atoms not involved in chain formation are omitted for clarity. | ||
Both cocrystal components in Form I are in general positions, while in Form II the opa molecule lies on a twofold axis running through the keto group and the bipy molecule lies on an inversion centre. Therefore, conformations of components in the underlying supramolecular motif, and thus the conformation of the motif as a whole, differ in the two polymorphs, which can thus be denoted as conformational polymorphs,15 as opposed to supramolecular synthon polymorphs.8d,e While the differences in bipy conformations in the two polymorphs amount to rotation of one ring relative to the other (the torsion angle changes from 11.4° in Form I to 0° in Form II), a significantly different spatial arrangement is evident for the opa component (Fig. 2). It is worthwhile to compare the conformations of opa found herein and conformation of pa in pa·bipy cocrystals.9 As mentioned previously, in the pa·bipy system the acid assumes an elongated conformation. Even though opa could also assume such an elongated conformation,16 in the herein prepared cocrystals it rather adopts a twisted conformation which in turn develops the 1-D zig-zag chains with opa at the kinks of the chains. In Form I, two types of C–H⋯O interactions contribute to the crystal structure stabilization. The first one involves the keto oxygen atom of opa and the pyridine hydrogen atom in the ortho position, which could be considered more acidic due to the proximity of the nitrogen atom. The second one concerns the hydrogen atom in the meta position and the oxygen atom of the carboxyl group (Fig. S5†). In all three polymorphs of the pa·bipy system, oxygen atoms of both carboxylic groups of pa are involved in C–H⋯O interactions exclusively towards bipy.8 In the structure of the Form I, the keto group replaces one of the carboxylic groups in interactions towards bipy.
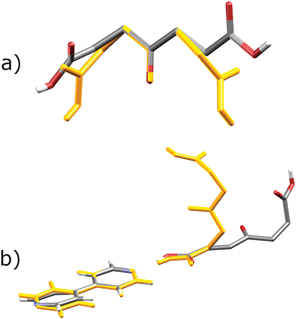 | ||
| Fig. 2 (a) Comparison of opa conformations in Form I (coloured) and Form II (orange). Molecules were overlapped by finding the best overlap of the two keto groups. (b) Twisting of the conformation of the supramolecular building block in hydrogen-bonded chains in Form I (coloured) and Form II (orange). | ||
On the other hand, the conformation of opa in the Form II is such that the keto group is sterically hindered by both carboxylate groups and is thus not involved in hydrogen bonding or any other short contacts. Instead, both carboxyl groups act as acceptors in the formation of crystallographically independent but chemically very similar C–H⋯O interactions towards neighbouring bipy and opa molecules (Fig. 3 and Fig. S6†).
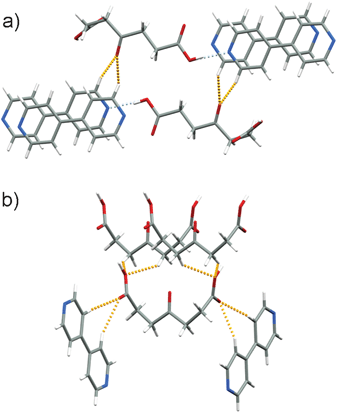 | ||
| Fig. 3 C–H⋯O interactions (dashed and orange) between hydrogen-bonded chains in (a) Form I and (b) Form II. | ||
Based on the previous discussion, it can be concluded that the keto group of the opa molecule acts only on the level of weak interactions but with a profound influence on the overall conformation of the hydrogen-bonded chain and packing in the resulting polymorphs. Its influence could be either direct when it is itself engaged in weak interactions or indirect when the changes in crystal packing result from changes in electrostatic properties of the molecule. However, it is worth pointing out that, in spite of the rather large conformational changes, the main supramolecular synthon persisted, which provides evidence of its robustness.
When compared to the spectra of pure opa and bipy, the IR spectra of Form I and Form II reflect differences in intermolecular forces stabilising structures of opa·bipy cocrystal polymorphs. The carboxylic group of opa gives characteristic absorptions which change noticeably if involved in different hydrogen bonding patterns.17Carboxylic acids exist predominantly as hydrogen-bonded dimers (synthon B, Scheme 1) in their condensed phases. The O–H stretching absorption band for such a symmetric R22(8) hydrogen bond is very strong and broad, extending over 600 cm−1. In opa it is visible from 3529–2520 cm−1 (ESI†). Additionally, weak peaks at 2666 cm−1 and 2528 cm−1 which are typical of such dimers also appear. In both forms of opa·bipy, disappearance of these weak bands and emergence of a broad band at ∼2450 cm−1 are assigned to stretching vibrations of O–H group involved in the O–H⋯N hydrogen bond (synthon A). The carbonyl group stretching frequency in pure opa is found at 1692 cm−1. In the spectrum of Form I it is increased by almost 30 cm−1, exhibiting a strong doublet (1719 cm−1 and 1700 cm−1). In Form II, the same band is found as a strong singlet at 1702 cm−1. Also, the C–O stretch, the O–H in-plane and the O–H out-of-plane deformation bands characteristic of the carboxylic acid dimer in opa (at 1428 cm−1, 1329 cm−1 and 904 cm−1, respectively18) are not observed in spectra of either of the polymorphs of the opa·bipy cocrystal. Strong bands at 1598 cm−1 in the spectrum of Form I and at 1600 cm−1 in Form II belong to the C![[partial double bond, top dashed]](https://www.rsc.org/images/entities/char_e12f.gif) N stretching vibrations of bipy (1590 cm−1 in pure bipy). Position of this band is a good indicator of whether the proton transfer between opa and bipy has occurred, since in pyridinium salts this band is located at ∼1630 cm−1.
N stretching vibrations of bipy (1590 cm−1 in pure bipy). Position of this band is a good indicator of whether the proton transfer between opa and bipy has occurred, since in pyridinium salts this band is located at ∼1630 cm−1.
The relative stabilities of the polymorphs and their solid-state interconversions were investigated by means of differential scanning calorimetry (DSC) and mechanochemical methods. DSC measurements in an inert atmosphere revealed a weakly endothermic peak at 57 °C (Fig. 4) upon heating of Form II. Powder diffraction experiment of the material, collected immediately after this thermal event occurred, clarified that it corresponded to the phase transition to Form I (ESI†). Consequently, the strong endotherm found at 151 °C (onset) is assigned to melting of the cocrystal and is the same regardless of which form was used for the experiment. No mass loss was observed during the phase transition and melting. Even though the system is enantiotropic,19 the reversible solid-state transformation did not occur upon cooling to −90 °C. Furthermore, rapid cooling of the melt resulted in formation of Form II.
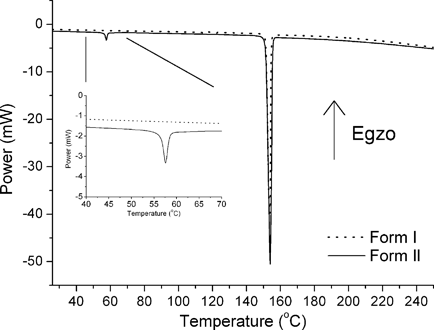 | ||
| Fig. 4 DSC traces of polymorphs of the opa·bipy cocrystal. The inset shows the thermal event corresponding to the transformation of Form II to Form I. A bigger endothermic peak corresponds to melting. | ||
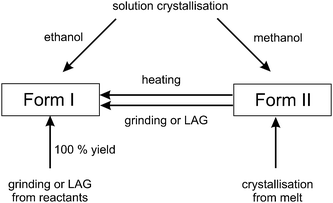
Solid-state grinding or LAG (with methanol or ethanol) of opa and bipy in a ball mill yielded exclusively Form I in quantitative yield. Also, Form II transforms to Form I upon short periods of grinding, while Form I was stable to grinding (Scheme 2).
 | ||
| Scheme 2 | ||
To summarise, we have isolated and characterised two conformational polymorphs of the cocrystal of opa and bipy. As expected, considering the robustness of the carboxylic acid-pyridine supramolecular synthon, both polymorphs consist of the same underlying 1-D hydrogen-bonded chains, as is found in polymorphs of pa·bipy cocrystal. However, the presence of the keto group in opa changes the conformation of the acid from elongated to twisted, resulting in kinked hydrogen-bonded chains. The described polymorphs demonstrate how large an effect weak interactions (mostly C–H⋯O hydrogen bonds) can have on the conformation of the basic supramolecular motif and thus on the overall crystal packing.
The solid-state transformation of Form II to Form I can be achieved either by thermal treatment or by mechanochemical methods. Additionally, solid-state synthesis of Form I from the reactants has been accomplished in 100% yield, by both grinding and liquid-assisted grinding. These results add to the growing interest in transformations and syntheses in the solid-state, which are considered to be environmentally friendly, since solvents are either entirely excluded or are present in only minimal amounts.
Acknowledgements
This work was financially supported by the Ministry of Science, Education and Sports of the Republic of Croatia (Grant Nos. 0119630 and 119-1191342-1082).References
- (a) C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC; (b) P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499 CrossRef; (c) G. R. Desiraju, CrystEngComm, 2003, 5, 466 RSC; (d) J. D. Dunitz, CrystEngComm, 2003, 5, 506 RSC; (e) A. D. Bond, CrystEngComm, 2007, 9, 833 RSC.
- (a) P. Vishweshwar, J. A. McMahon and M. J. Zaworotko, Crystal Engineering of Pharmaceutical Co-crystals, in Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. J. Vittal, Wiley, Chichester, UK, 2006, pp. 25–49 Search PubMed; (b) S. Karki, T. Friščić, L. Fábián, P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905 CrossRef CAS; (c) D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 2252 CrossRef CAS; (d) A. V. Yadav, A. S. Shete, A. P. Dabke, P. V. Kulkarni and S. S. Sakhare, Indian J. Pharm. Sci., 2009, 71, 359–370 Search PubMed; (e) A. V. Trask, S. W. D. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013 CrossRef CAS; (f) D. Braga and F. Grepioni, Chem. Commun., 2005, 3635 RSC; (g) Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889 RSC; (h) S. Aitipamula, P. S. Chowa and R. B. H. Tan, CrystEngComm, 2009, 11, 1823 RSC; (i) N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950 CrossRef CAS; (j) S. L. Childs and K. I. Hardcastle, Cryst. Growth Des., 2007, 7, 1291 CrossRef CAS.
- (a) D. Cinčić, T. Friščić and W. Jones, Chem. Mater., 2008, 20, 6623 CrossRef CAS; (b) L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS; (c) A. Matsumoto, Polym. J., 2003, 35, 93 CrossRef CAS; (d) L. R. MacGillivray, J. Org. Chem., 2008, 73, 3311 CrossRef CAS; (e) R. Custelcean and J. E. Jackson, J. Am. Chem. Soc., 1998, 120, 12935 CrossRef CAS.
- (a) C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397 RSC; (b) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS; (c) A. N. Sokolov, D.-K. Bučar, J. Baltrusaitis, S. X. Gu and L. R. MacGillivray, Angew. Chem., Int. Ed., 2010, 49, 4273 CrossRef CAS.
- (a) C. B. Aakeröy, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 569 CrossRef; (b) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (c) D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1 RSC.
- (a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS; (b) C. B. Aakeröy, J. Desper and B. M. T. Scott, Chem. Commun., 2006, 1445 RSC; (c) T. Friščić and W. Jones, Faraday Discuss., 2007, 136, 167 RSC; (d) M. Du, Z.-H. Zhang and X.-J. Zhao, Cryst. Growth Des., 2005, 5, 1199 CrossRef CAS; (e) A. Dey, M. T. Kirchner, V. R. Vangala, G. R. Desiraju, R. Mondal and J. A. K. Howard, J. Am. Chem. Soc., 2005, 127, 10545 CrossRef CAS; (f) D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932 CrossRef CAS; (g) D. Cinčić, T. Friščić and W. Jones, New J. Chem., 2008, 32, 1776 RSC.
- C. V. K. Sharma, Cryst. Growth Des., 2002, 2, 465 CrossRef CAS.
- (a) G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007 CrossRef CAS; (b) M. Du, X.-J. Jiang, X. Tan, Z.-H. Zhang and H. Cai, CrystEngComm, 2009, 11, 454 RSC; (c) B. R. Bhogala, S. Basavoju and A. Nangia, CrystEngComm, 2005, 7, 551 RSC; (d) N. J. Babu, L. S. Reddy, S. Aitipamula and A. Nangia, Chem.–Asian J., 2008, 3, 1122 CrossRef CAS; (e) B. R. Sreekanth, P. Vishweshwar and K. Vyas, Chem. Commun., 2007, 2375 RSC; (f) D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, T. B. Borchardt, L. R. MacGillivray and G. G. Z. Zhang, Mol. Pharmaceutics, 2007, 4, 339 CrossRef CAS.
- D. Braga, G. Palladino, M. Polito, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem. Eur. J., 2008, 14, 10149 CrossRef CAS.
- D. Braga, S. L. Giaffreda, M. Curzi, L. Maini, M. Polito and F. Grepioni, J. Therm. Anal. Calorim., 2007, 90, 115 CrossRef CAS.
- (a) T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621 CrossRef CAS; (b) N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372 RSC; (c) D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 1106 CrossRef CAS; (d) S. Karki, T. Friščić and W. Jones, CrystEngComm, 2009, 11, 470 RSC; (e) A. V. Trask and W. Jones, Top. Curr. Chem., 2005, 254, 41 CAS; (f) T. Friščić, J. Mater. Chem., 2010, 20, 7599 RSC.
- Search parameters: CSD version 5.31, November 2009 (2 updates), organics only, compounds that contain pyridine and carboxyl; no pyridinium, no metal.
- Initial search modified by including hydrogen atoms on pyridine 2,6-positions. 442 entries found containing both pyridine and carboxyl groups. Synthon A in 389 entries or 88%; synthons B and C in only 3 and 4 entries, respectively.
- (a) T. Steiner, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 103 CrossRef CAS; (b) F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25 RSC; (c) T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533 CrossRef CAS; (d) S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2009, 9, 2881 CrossRef CAS.
- (a) J. Bernstein and T. A. Hagler, J. Am. Chem. Soc., 1978, 100, 673 CrossRef CAS; (b) L. Yu, G. A. Stephenson, C. A. Mitchell, C. A. Bunnell, S. V. Snorek, J. J. Bowyer, T. B. Borchardt, J. G. Stowell and S. R. Byrn, J. Am. Chem. Soc., 2000, 122, 585 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425 CrossRef.
- (a) B. A. Dawa and J. A. Gowland, Can. J. Chem., 1978, 56, 2567 CrossRef CAS; (b) G. M. Florio, T. S. Zwier, E. M. Myshakin, K. D. Jordan and E. L. Silbert III, J. Chem. Phys., 2003, 118, 1735 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Frequencies of Organic Molecules, Academic Press, San Diego, 1991, pp. 138–141 Search PubMed.
- F. H. Herbstein, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 341 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. CCDC reference numbers 785101 and 785102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0nj00569j |
| ‡ aCrystal data for Form I: C7H10O5·C10H8N2, Mr = 330.33, monoclinic, a = 11.3107(10) Å, b = 9.4127(8) Å, c = 15.8812(13) Å, α = 90.00°, β = 102.668(7)°, γ = 90.00°, V = 1649.6(2) Å3, T = 293(2) K, space groupP21/n, Z = 4, μ(MoKα) = 0.099 mm−1, 15846 reflections measured, 2900 independent reflections (Rint = 0.0274). Final values: R1, 0.0502 (I > 2σ(I)); wR(F2), 0.1381 (I > 2σ(I)); R1, 0.0627 (all data); wR(F2), 0.1490 (all data). Goodness of fit on F2 = 1.162. Crystal data for Form II: C7H10O5·C10H8N2, Mr = 330.33, monoclinic, a = 7.583(3) Å, b = 8.752(2) Å, c = 24.527(6) Å, α = 90.00°, β = 92.29(3)°, γ = 90.00°, V = 1626.5(8) Å3, T = 293(2) K, space groupC2/c, Z = 4, μ(MoKα) = 0.100 mm−1, 2479 reflections measured, 1579 independent reflections (Rint = 0.0305). Final values: R1, 0.0569 (I > 2σ(I)); wR(F2), 0.1599 (I > 2σ(I)); R1, 0.0742 (all data); wR(F2), 0.1800 (all data). Goodness of fit on F2 = 1.088. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |