Multifunctional ligands in transition metal catalysis
Robert H.
Crabtree
Yale University Chemistry Department, 225 Prospect St., New Haven, CT 06520, USA
First published on 24th November 2010
Abstract
Sophisticated ligands are now being designed that do far more than just fulfil their traditional spectator roles by binding to the metal and providing a sterically-defined binding pocket for the substrate in homogeneous transition metal catalysis. This Focus review emphasizes selected cases in which ligands carry additional functional groups that change the properties of the ligand as a result of an external stimulus or undergo catalytically-relevant ligand-based reactivity. These include proton responsive ligands capable of gaining or losing one or more protons, ligands having a hydrogen bonding function, electroresponsive ligands capable of gaining or losing one or more electrons, and photoresponsive ligands capable of undergoing a useful change of properties upon irradiation. Molecular recognition ligands and proton coupled electron transfer (PCET) are briefly discussed.
 Robert H. Crabtree | Bob Crabtree was educated at New College, Oxford with Malcolm Green, did his PhD with Joseph Chatt at Sussex and spent four years in Paris with Hugh Felkin at the CNRS, Gif. At Yale since 1977, he is now Whitehead Professor. He has been an ACS and RSC organometallic chemistry awardee, Baylor Medallist, Mond lecturer, Kosolapoff awardee, Stauffer lecturer, has chaired the ACS Inorganic Division and is the author of an organometallic textbook. Early work on catalytic alkane C–H activation and functionalization was followed by work on H2 complexes, dihydrogen bonding, and catalysis for green and energy chemistry. His homogeneous hydrogenation catalyst is now widely used. |
Introduction
In addition to the traditional ligand functions of binding to a metal and provision of steric hindrance or of bridging groups, the title ligands have one or more additional functions that can be useful in catalysis. In the current state of the field, these functions include:1. proton responsive ligands capable of undergoing a change of properties on gaining or losing one or more protons;
2. ligands having a hydrogen bonding function, implying partial proton transfer to or from a suitable partner;
3. electroresponsive ligands capable of undergoing a change of properties on gaining or losing one or more electrons;
4. photoresponsive ligands capable of undergoing a change of properties on irradiation;
5. ligands having a molecular recognition function; and
6. cases that fall outside the scope of the present article including polymer- and biopolymer-based ligands and metalloenzymes, which provide by far the most sophisticated multifunctional ligands in the shape of the polypeptide component of the protein.
This list will no doubt grow in future as further examples are identified.
By gaining or losing protons, proton responsive ligands are transformed into ones that are lesser or greater donors, allowing access to a wider range of ligand properties than can be provided by a classical unresponsive spectator ligand. If, during the transformation of the substrate in the catalytic reaction, the substrate has to gain or lose protons, such ligands can in principle also aid proton management by mediating proton transfer to and from the substrate. Hydrogen bonding ligands form a closely related group in that a hydrogen bond is essentially an incomplete proton transfer.
By gaining or losing electrons, electroresponsive ligands not only access different ligand properties from the same structure, but can in principle also participate in multi-electron reactions, one electron transferring to or from the metal and one transferring to or from the ligand. This is significant because in moving from the platinum group metals (PGMs) to the much less expensive first row transition metals, we move from metals that naturally undergo two electron redox processes to ones that prefer one electron processes. This is illustrated, for example, by the triad of oxidation states normally available to iridium vs. cobalt—Ir(I),(III) and (V) vs. Co(I),(II) and (III). Thus, if the ligand and metal can combine their redox powers, multi-electron processes can in principle be accessible, even to first row metals. Indeed, Chirik and Wieghardt argue that redox active ligands may be able to endow base metals with high catalytic activity of the PGMs and thus “confer nobility on base metal catalysts”.1
A ligand is expected to become a poorer sigma donor and/or a better pi acceptor if it is protonated because of the increase in the positive charge or the decrease in the negative charge on the ligand. For example, the NH2 ligand in M–NH2 is a strong pi donor via both its pi and sigma lone pairs, while NH3 in M–NH3 is a weaker sigma donor and lacks pi donor power. Likewise, ligand oxidation also decreases donor power by decreasing the negative charge or increasing the positive charge on the ligand.
Electron and proton responsive ligands can both undergo either internal or external transfer of electrons or protons. Some cases will be discussed below where a proton is transferred from one ligand to another or an electron is transferred between metal and ligand in an internal transfer. In other cases, the transfer occurs to or from an external reactant or electrode.
Molecular recognition ligands have been devised that achieve a high specificity by combining molecular recognition2 with chemical catalysis, just as enzymes do in biology.3 With a looser definition of molecular recognition, ligands for asymmetric catalysis4 could even be considered as falling into this category in the sense that the asymmetric ligand creates an asymmetric cavity for recognition of the transition state for the reaction. These asymmetric binding pockets involve repulsive interactions, however, and therefore do not enter into the full spirit of molecular recognition, a term that implies the intervention of attractive interactions. Good reviews are in any case available for this topic,5 so we will not discuss such ligands here.
The very diverse sixth group, not covered in detail here, includes ligands that permit easy separation from a reaction mixture for catalyst recycling.6 Some of these impart specific physical properties, such as fluorous ligands that cause the catalyst to separate spontaneously from the reaction mixture.7 More exotic ligands include dendrimer functions to allow catalyst separation by nanofiltration.8 Numerous ligands have been equipped with anchor functions that allow attachment to a surface, again for catalyst recycling.9 Specialized ligands have been increasingly used to impart useful photophysical properties that go far beyond catalysis. Examples include iridium complexes for use in organometallic light emitting diodes10 and ruthenium complexes as sensitizers for Graetzel solar cells.11 In this case, the dipyridyl or terpyridyl ligands of the ruthenium complex sensitizer are equipped with carboxylic acid groups that permit attachment to the TiO2 nanoparticulate electrodes commonly used in such cells. Other ligands have been designed to form supramolecular constructs such as catenanes12 and metal–organic frameworks.13 Rather than being a detailed review of each class of multifunctional ligand, this Focus article serves to draw attention to the relationship between these ligand classes.
Proton responsive ligands
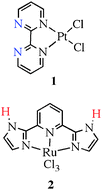
The work of Periana and co-workers14 on CH activation in general, and on methane-to-methanol conversion in particular, provides an excellent illustration of the use of this type of ligand in an important catalytic reaction. In his work in strong acid media, the role of the acid solvent is to capture the product methanol, either by protonation or by formation of bisulfate ester, thus preventing over-oxidation, ultimately to carbon dioxide. The ligand they employ, shown as 1 nearby, is not only capable of binding to the metal but also of binding up to two protons at the free nitrogen lone pairs of the bipyrimidine, shown in blue in the diagram. This not only has the effect of modifying the ligand properties by enhancing its pi donor power, but also prevents loss of the ligand from the metal in the harsh acid environment, as occurs with analogous ligands lacking proton acceptor sites.In more recent work from the same group, the reverse strategy has been employed. Instead of acid, strong base is employed as the medium for CH activation reactions. In this case, the ligand chosen, shown as 2 in the graphic, has labile protons, shown in red, that can be removed in the strongly basic environment. This deprotonation has the effect of enhancing the donor power of the ligand and accelerating the rate of CH activation in the system.15
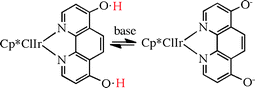
Himeda and co-workers16 have provided another striking example of a ligand undergoing deprotonation in basic media and thus promoting a higher catalytic activity in the metal complex to which it is attached; in this case, the reaction is carbon dioxide reduction to formic acid with hydrogen. As a bonus, at the end of the reaction, the ligand is reprotonated with the result that the catalyst precipitates and can be recycled (eqn (1)).
 | (1) |
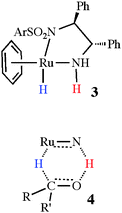
Noyori and co-workers17 have introduced an extremely successful asymmetric catalyst for the transfer hydrogenation of ketones. The point of interest for the present discussion is that the active catalyst, 3, contains a nucleophilic hydride (in blue) and an adjacent ligand-based electrophilic proton (in red), which are believed to be transferred in a concerted fashion to the substrate, as shown in the proposed transition state, 4. In the catalytic cycle, the hydride and proton are reinstalled by reaction of the depleted catalyst with the primary reductant, often a mixture of NEt3 and formic acid.
Numerous proton responsive ligands are known. Milstein and co-workers18 have an attractive catalyst for a ‘green’ synthesis of amide RCONHR′ from alcohol RCH2OH and amine R′NH2via dehydrogenation of hemiaminal intermediate RCH(OH)NHR′ by a Ru complex of a PNN pincer ligand. During the reaction, deprotonation of the pincer ligand occurs in the basic medium, and this is considered to be a key step in the mechanism. DuBois, Rakowski-DuBois and co-workers19 have reported an iron hydride containing a PNP ligand, Et2PCH2N(CH3)CH2PEt2, that chelates via the P atoms, allowing the uncoordinated central N atom to act as a pendant base. In this case, protonation at this nitrogen cannot significantly affect the donor characteristics of the ligand. Instead, the protonated nitrogen mediates hydride/proton exchange via an intermediate dihydrogen complex, formed by reversible proton transfer from the pendant NH group to the hydride ligand.
Hydrogen bonding ligands
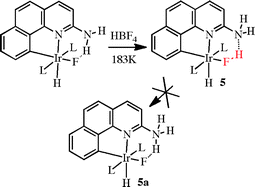
Hydrogen bonding is intimately involved in the catalytic process in organocatalysis, but its involvement in organometallic catalysis is still limited. Nevertheless, interesting effects have been seen in stoichiometric ligand binding and H–H activation. Hydrogen bonding is also involved in molecular recognition catalysis, discussed in a separate section below.Hydrogen bonding ligands can assist a metal in binding a substrate when their hydrogen bonding groups are able to interact with substrates that are simultaneously bound to the metal. For example, the hydrogen fluoride complex shown in complex 5 in eqn (2) is only accessible when the ligand contains a hydrogen bonding amine group (L = PPh3).20 That the complex is truly one of hydrogen fluoride, as in 5, and not simply of fluoride ion hydrogen-bonded to an N–H group, as in 5a, was shown by the high value of the proton–fluorine coupling in the NMR spectrum of the complex (1JHF = 440 Hz), a value characteristic of a hydrogen-bonded N⋯H–F structure.
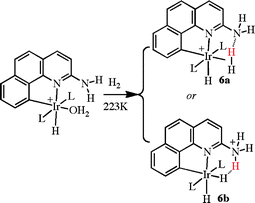
A hydrogen bonding ligand can also mediate proton transfer, blurring the distinction between the proton responsive and hydrogen bonding classes of ligand. In a related case, for example, the formation of a dihydrogen complex leads to proton transfer from the hydrogen molecule to the adjacent amine group, as shown in eqn (3).21 Both structures, with (6b) and without (6a) proton transfer to nitrogen, were identified in a series of complexes bearing different ligands (e.g., L = PCy3 or PPh3). The controlling factor proves to be ion pairing, possible for the dihydrogen complex only with ligands that are relatively smaller in size, such as PPh3, but not with the bulkier ligand PCy3. Only the smaller ligands permit the anion PF6− to approach close enough to the metal to allow ion pairing with the H2 ligand. The bulky PCy3 ligand causes the anion to retreat to a more distant location, causing the stabilization of structure 6b, since ion pairing is now only sterically possible at the remote Ar–NH3+ site. Eqn (3) certainly constitutes an internal proton transfer, and even in eqn (2), initial protonation at nitrogen prior to proton transfer to fluoride is quite likely.
 | (2) |
 | (3) |
Electron responsive ligands
Non-innocent ligands—ones that can adopt more than one oxidation state—can make it difficult to assign a specific oxidation state to the metal. For example, Gray and co-workers proposed that a series of neutral cobalt and nickel bis-dithiolene complexes were better described as M(II) compounds, with each ligand having radical anion character (7a), rather than as an M(IV) species with conventionally dianionic dithiolenes (7b).22 Numerous more recent examples of non-innocent ligands exist.23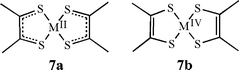
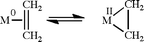
In hydrogenation catalysis, molecular hydrogen might be considered as non-innocent, but only in the sense that it can convert from a dihydrogen complex to a dihydride form, formally oxidizing the metal by two units, as first seen very early in the development of the field of dihydrogen complexes with the Kubas complex itself (eqn (4)). The same internal redox reaction holds for any ligand that can undergo a reversible oxidative addition from a sigma complex, such as a C–H bond in a reversible cyclometalation or an equilibrium between a silane sigma complex and a silyl hydride (eqn (5)).24 An analogous situation exists for ethene, C2H4, where metalacyclopropane and Chatt–Dewar extreme formulations are possible, depending on the degree of electron transfer from metal to ligand (eqn (6)).25 Once again, all of these constitute internal electron transfers.
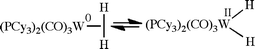 | (4) |
 | (5) |
 | (6) |
More interesting are cases where an external electron transfer causes a ligand to modify its characteristics, typically its donor power to the metal, thus allowing access to a number of different states of the ligand. Early examples of such external redox active ligands again include dithiolenes.26 Stiefel and co-workers have provided an elegant example, in which changing the oxidation state of a nickel dithiolene complex causes a large change in the properties of the complex as a whole.27 With the goal of separating C2H4 from a stream of mixed gases, these authors oxidized [NiL2]− to [NiL2] (L = (CF3CS)2). The resulting neutral complex reacts with C2H4, not at the metal but at the ligand, as shown in eqn (7). Subsequent reduction to the anionic form releases C2H4. Remarkably, the system is not poisoned by C2H2, H2, CO or H2S, gases which are all commonly present in industrial gas streams, no doubt because C2H4 is the only one of these gases that is able to bind to the sulfur ligand. This also illustrates an additional type of ligand function: direct reaction of a substrate or reactant at a ligand.
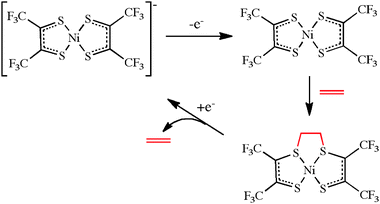 | (7) |
Proton transfer and electron transfer to or from ligands can also be concerted, as in the proton coupled electron transfer process (PCET). This net hydrogen atom transfer is important in catalytic water oxidation,28 artificial photosynthesis29 and in a wide variety of enzymes.30 For example, removal of a proton and an electron via PCET from an Mn(III) aqua complex twice over would give a formally Mn(V) oxo complex of a type typically proposed as an intermediate in water oxidation in photosynthesis (eqn (8)).31
| LnMnIII(OH2) ⇒ LnMnVO + 2(H+ + e−) | (8) |
Photoresponsive ligands
Although commonly employed in molecular switches and related devices,32 and in supramolecular catalysis,33 photoresponsive ligands constitute a surprising gap in the roster of functional ligands so far examined for homogeneous transition metal catalysis. Azobenzene cis–trans photoisomerism is an attractive target for initial studies, and a few reports have appeared. An attempt by Kudo and co-workers to influence asymmetric allylic alkylation catalysis by irradiation of a palladium complex of ligand 8 with 354 nm light proved unavailing because no change of ee was seen.34 Segarra-Maset et al. have successfully induced cis–trans isomerism of the azobenzene group in ligand 9 with 350 nm light, but this very recent work has not yet been followed up with catalytic applications.35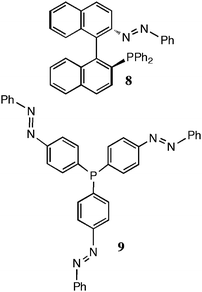
Branda and co-workers have seen substantial irradiation-dependent changes in the ee and dr values in the catalytic cyclopropanation of styrene with the copper(I) complex of 10a/10b. The open and closed forms of the ligand are photoswitchable with different wavelengths of light, as indicated in eqn (9).36
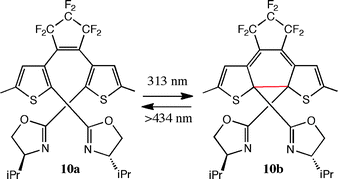 | (9) |
Molecular recognition ligands
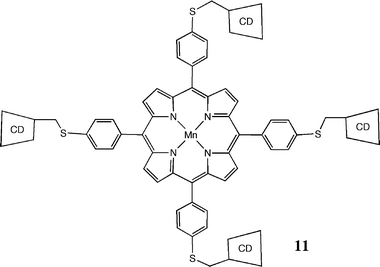
Lehn's review from 199037 details a vast array of applications of molecular recognition, many of which involve multifunctional ligands as well as some supramolecular catalysis, but transition metal catalysis was not a major contributor at that time. New reports that bring the topic up to the present time have appeared from Meeuwissen and Reek,38 and Prins and co-workers.39 We will not attempt to duplicate their coverage but just mention some typical cases.In Breslow's early porphyrin complex (11) beta-cyclodextrins (CD in the diagram) play a molecular recognition role by holding the substrate for reaction at a specific site. On hydroxylation with PhIO as the primary oxidant, the 6-hydroxy derivative of the cholestanol substrate was formed by reaction with a formally Mn(V) oxo intermediate. The substrate was a cholestanol modified with two hydrophobic t-butylphenyl binding groups to interact with the cyclodextrin and additional sulfonic acid groups to enhance substrate solubility.40
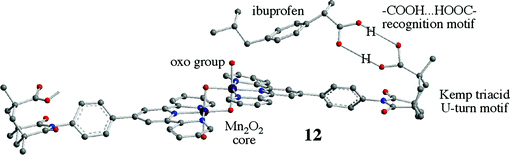
A more recent example is our own work in collaboration with Brudvig on complex 12.41 In this case, the ligand binds to a manganese oxo cluster via a terpyridine function and to the substrate for the catalytic reaction via the remote carboxylate function by –COOH⋯HOOC– hydrogen bonding. The substrate, in this case the drug ibuprofen, is held in such a way that a specific CH bond is held close to the reactive oxo group, such that hydroxylation takes place at that site remote from the binding group. In the case of 4-methylcyclohexane acetic acid as the substrate, the reaction takes place with a defined regio- and stereoselectivity. The primary oxidant in this case is the O atom donor [O–OSO3]2−, and the reaction is believed to proceed via a formally Mn(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate, although computational work suggests the active spin isomeric form is best described as an Mn(IV)–O˙ oxyl. Both Mn complexes 11 and 12 are believed to follow a ‘rebound’ mechanism, in which the oxo or oxyl abstracts a hydrogen atom from the C–H bond to give a C-centered radical and a Mn(IV)–OH intermediate. The C radical then abstracts a hydroxyl radical from the Mn(IV)–OH to form the C–OH carbinol and regenerate the Mn(III) catalyst.
O intermediate, although computational work suggests the active spin isomeric form is best described as an Mn(IV)–O˙ oxyl. Both Mn complexes 11 and 12 are believed to follow a ‘rebound’ mechanism, in which the oxo or oxyl abstracts a hydrogen atom from the C–H bond to give a C-centered radical and a Mn(IV)–OH intermediate. The C radical then abstracts a hydroxyl radical from the Mn(IV)–OH to form the C–OH carbinol and regenerate the Mn(III) catalyst.
The self-assembly of ligands by molecular recognition has also received attention. As early as 1978, Whitesides and co-workers42 formed an asymmetric ligand by the self-assembly of a chiral avidin protein with an achiral biotin-functionalized phosphine. The Rh(I) complex proved to be an asymmetric hydrogenation catalyst for an acetamidoacryclic acid with ee values of 40%.
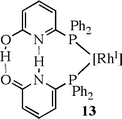
Breit and co-workers43 formed an unsymmetrical chelating diphosphine from the self-assembly of a heterodimer of a tautomeric pair of monodentate 2-pyridone and 2-hydroxypyridine-substituted phosphines. This complex (13) proved very effective in hydroformylation.
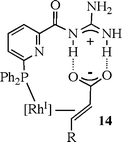
The same group has recently assembled a more elaborate ligand in which an α,β-unsaturated carboxylic acid substrate is oriented by a guanidine group appended to the Rh-bound phosphine, providing regiocontrol in the hydroformylation. As shown in structure 14, proton transfer from the acid to the guanidine provides an ion pair that enhances the hydrogen bonding interaction.44
Ligand- vs. metal-based properties?
Up to now, we have implicitly assigned all the special properties described above to the ligand. In fact, the full complex is always responsible, and assignment of the effect to the ligand or to the metal may oversimplify the situation. In this context, no ligand is completely innocent—even simple ligands must participate to some extent in what is conventionally assigned as a metal-based redox process, for example.In some cases, the ligand is arguably the predominant contributor to the multifunctional property, such as in the hydrogen bonding ligands 5 and 6, the photoresponsive ligands 8–10, and the molecular recognition ligands 11, 12 and 14. In others, the property arises from a cooperative effect between metal and ligand, as in the Noyori outer sphere hydrogenation in complex 3, in the electronic structure of dithiolene complex 7 and in the Stiefel reaction of eqn (7). Each case therefore needs to be critically examined on its own merits.
The proposed role of any additional functional group on a ligand also needs to be tested. Where possible, a useful procedure is to carry out a control study on a ligand that is as close as possible to the real one, but in which the additional function is suppressed. For example, one could replace the 2,2′-dipyrimidyl ligand in 1 with 2,2′-dipyridyl or the bis-imidazole in 2 with the bis-N-methyl imidazole analogue. Metal reduction potentials are also a useful indication, since these are very sensitive to the ligand environment.45
Future directions that the field might take include the provision of Lewis acid sites in the vicinity of the metal. Indeed, a recent result in this direction comes from a paper by Miller et al.46 on an L2Re(CO)4 complex bearing a Ph2PCH2CH2BR2 ligand. Dihydrogen activation at the boron center is achieved in the presence of a base (frustrated Lewis pair effect). The resulting Ph2PCH2CH2BHR2 intermediate can then transfer hydride to the coordinated CO groups.
Conclusion
Multifunctional ligands have important past, present and prospective roles in catalysis, but the variety of possible structures awaiting design and synthesis is so large that we are only limited by our imagination in introducing novel and useful functions into homogeneous transition metal catalysis.Acknowledgements
I gratefully thank Gary Brudvig and Odile Eisenstein, both long-standing collaborators in catalysis, for many illuminating discussions, as well as the Department of Energy, Office of Basic Energy Sciences, for generous support of our work under the catalysis, solar and EFRC programs with grants DE-FG02-84ER13297, DE-FG02-07ER15909, DE-PS02-08ER15944, DE-SC-0001055 and DE-SC-0001298.References
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304 CrossRef.
- Enzyme Kinetics and Mechanism, ed. P. F. Cook and W. W. Cleland, Garland Science, London/New York, 2007 Search PubMed.
- R. Noyori, Adv. Synth. Catal., 2003, 345, 15 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS and references cited therein.
- J. A. Gladysz, Chem. Rev., 2002, 102, 3215 CrossRef CAS.
- I. T. Horvath and J. Rabai, Science, 1994, 266, 72 CrossRef CAS.
- R. van de Coevering, A. P. Alfers, J. D. Meeldijk, E. Martinez-Viviente, P. S. Pregosin, R. J. M. K. Gebbink and G. van Koten, J. Am. Chem. Soc., 2006, 128, 12700 CrossRef CAS.
- C. E. Song and S. G. Lee, Chem. Rev., 2002, 102, 3495 CrossRef CAS.
- S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923 CrossRef CAS.
- A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef CAS.
- M. Beyler, V. Heitz and J. P. Sauvage, New J. Chem., 2010, 34, 1825 RSC.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58 CrossRef CAS.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS.
- B. G. Hashiguchi, K. J. H. Young, M. Yousufuddin, W. A. Goddard and R. A. Periana, J. Am. Chem. Soc., 2010, 132, 12542 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS.
- R. M. Henry, R. K. Shoemaker, D. L. DuBois and M. Rakowski-DuBois, J. Am. Chem. Soc., 2006, 128, 3002 CrossRef CAS.
- D. H. Lee, H. J. Kwon, P. P. Patel, L. M. Liable-Sands, A. L. Rheingold and R. H. Crabtree, Organometallics, 1999, 18, 1615 CrossRef CAS.
- K. Gruet, E. Clot, O. Eisenstein, D. H. Lee, B. Patel, A. Macchioni and R. H. Crabtree, New J. Chem., 2003, 27, 80 RSC.
- E. Billig, R. Williams, I. Bernal, J. H. Waters and H. B. Gray, Inorg. Chem., 1964, 3, 663 CrossRef CAS.
- M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275 RSC; M. R. Ringenberg, S. L. Kokatam, Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 788 CrossRef CAS.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, New York, 5th edn, 2009 Search PubMed.
- N. Robertson and L. Cronin, Coord. Chem. Rev., 2002, 227, 93 CrossRef CAS.
- E. I. Stiefel and K. Wang, Science, 2001, 291, 106 CrossRef CAS.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727 CrossRef CAS.
- L. Hammarstrom and S. Styring, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1283 CrossRef.
- S. Hammes-Schiffer, E. Hatcher, H. Ishikita, J. H. Skone and A. V. Soudackov, Coord. Chem. Rev., 2008, 252, 384 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 3428 CrossRef CAS.
- Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001 Search PubMed.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2003, 125, 2224 CrossRef CAS.
- M. Kawamura, R. Kiyotake and K. Kudo, Chirality, 2002, 14, 724 CrossRef CAS.
- M. D. Segarra-Maset, P. W. N. M. van Leeuwen and Z. Freixa, Eur. J. Inorg. Chem., 2010, 2075 CAS.
- D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019 CrossRef.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304 CrossRef.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS.
- G. Gasparini, M. Dal Molin and L. J. Prins, Eur. J. Org. Chem., 2010, 2429 CrossRef CAS; A. Scarso, G. Zaupa, F. Bodar Houillon, L. J. Prins and P. Scrimin, J. Org. Chem., 2007, 72, 376 CrossRef CAS.
- R. Breslow, Y. Huang, X. Zhang and J. Yang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11156 CrossRef CAS; J. Yang, B. Gabriele, S. Belvedere, Y. Huang and R. Breslow, J. Org. Chem., 2002, 67, 5057 CrossRef CAS.
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941 CrossRef CAS.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS.
- B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608 CrossRef CAS.
- T. Smejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 3946 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271 CrossRef CAS.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2010, 132, 3301 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |