Substituted diethynyldithia[3.3]paracyclophanes—synthetically more accessible new building blocks for molecular scaffolding†
Jian-Long
Xia
,
Chan
Zhang
,
Xingxun
Zhu
,
Yaping
Ou
,
Guo-Jun
Jin
,
Guang-ao
Yu
and
Sheng Hua
Liu
*
Key Laboratory of Pesticide and Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: chshliu@mail.ccnu.edu.cn
First published on 8th October 2010
Abstract
A series of dialkyne building blocks based on the dithia[3.3]paracyclophane unit have been synthesized in good yields. The electronic properties of these novel dialkynes can be tuned through a transannular substitution effect. These synthetically more accessible diethynyldithia[3.3]paracyclophanes are promising candidates for the building of relatively carbon rich molecular scaffolds.
Introduction
The molecular building block associated with paracyclophanes is widely used in chiral catalysis,1 the design of new optoelectronic (NLO) materials,2polymer chemistry and materials science,3electron transfer processes,4 molecular electronics,5 novel nanoscale materials,6 and even organic solar cells.7 The unique structural and electronic properties of these derivatives result from the characteristic interactions between the two cofacial π-electron systems.8Acetylenic cyclophanes as a series of carbon-rich compounds not only show interesting and important structural and chemical behavior themselves, but also have been incorporated into ‘extended’ π-systems and polymers through coupling reactions such as the Sonogashira or Glaser couplings. Besides addition reactions, ethynyl cyclophanes can be employed to prepare novel iron, platinum and cobalt metal complexes.9a By polycondensation reactions, monomeric cyclophanes can be converted into novel polymers displaying lateral π-conjugation. Marrocchi, Taticchi7,9b–h and Hopf9i–k have done excellently work in this area.
Up to now, although the unique transannular π–π interaction associated with [2.2]paracyclophanes has been extensively studied and documented1,2,5,7,10 (Fig. 1), dithia[3.3]paracyclophane building blocks, which are synthetically more accessible, have received less attention.
![The structure of [2.2]paracyclophane and dithia[3.3]paracyclophane](/image/article/2011/NJ/c0nj00553c/c0nj00553c-f1.gif) | ||
| Fig. 1 The structure of [2.2]paracyclophane and dithia[3.3]paracyclophane | ||
In our opinion, the dithia[3.3]paracyclophane unit also represents a particularly valuable core system. It has been shown that the transannular effect in the [3.3]cyclophane unit is sufficiently strong to allow tuning of the optical properties of copolymers.11 In addition, dithia[3.3]paracyclophanes could be readily prepared by coupling reactions under high dilution conditions if different substituents on the cyclophane are desired.
We have now focused on the synthesis and studies of transannular π–π interactions in the dithia[3.3]paracyclophane unit. Here, we report the synthesis of a series of novel functionalized diethynyldithia[3.3]paracyclophane building blocks (9a–e, 10 and 11), as well as a preliminary study on the transannular substitution effect on their optical and electronic structures.
Results and discussion
Dithiaparacyclophanes 3a–e, 5a and 5b were prepared by coupling the corresponding pair of dithiol and dibromide under high dilution conditions according a modified procedure found in the literature (Scheme 1).11 The trimethylsilyl-protected bis-(ethynyl)ligand precursors 6a–e and 7 were synthesized by the Pd(PPh3)4/CuI-catalyzed Sonogashira cross-coupling reaction between 3a–e, 5a and trimethylsilylacetylene (Scheme 2). In the case of tetrafluoro substituents dibromo-dithiaparacyclophane 5b, however, this reaction did not proceed satisfactorily. Diethynyl precursor 8 was formed by the coupling of 5b and 2-methylbut-3-yn-2-ol under the same conditions. The ligand precursors were smoothly converted into the corresponding diethynyldithia[3.3]paracyclophanes 9a–e and 10 by removal of the trimethylsilyl protecting groups with dilute aqueous KOH in MeOH–THF, while 11 was obtained by treating 8 with NaH in toluene after refluxing for 2.5 h (Scheme 3).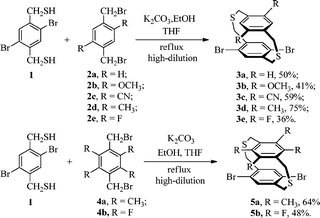 | ||
| Scheme 1 Synthetic route to 3a–e, 5a and 5b. | ||
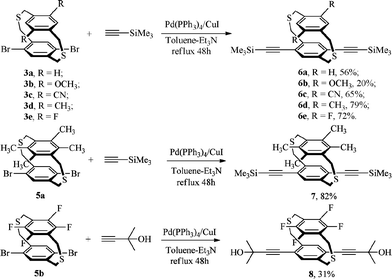 | ||
| Scheme 2 Synthesis of protected diethynyl precursors 6a–e, 7 and 8. | ||
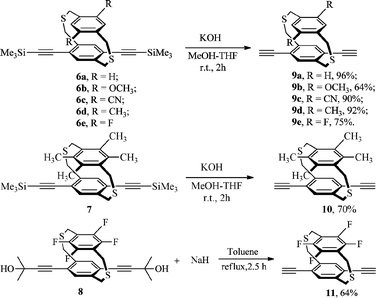 | ||
| Scheme 3 Synthesis of the dialkyne complexes 9a–e, 10 and 11. | ||
All of the synthetic compounds were characterized well by NMR and elemental analysis, and the molecular structure of 9a–e, 10 and 11 was also unambiguously supported by high resolution ESI mass spectra. The structure of 5b was determined by X-ray crystallography, and an ORTEP drawing of 5b is shown in Fig. 2.† The interplane distance between the two benzene rings of 3.258 Å is less than the normal packing distance of aromatic rings in organic aromatic molecules (3.4 Å), thus supporting a potential transannular π–π interaction between the two rings in the cyclophane unit.
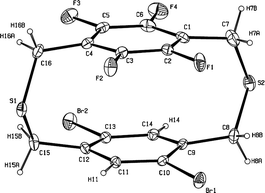 | ||
| Fig. 2 An ORTEP drawing of compound 5b with 20% probability ellipsoids. | ||
The electronic properties of diethynyldithia[3.3]-paracyclophanes 9a–e, 10 and 11 were investigated by absorption and emission spectroscopy (Table 1). The absorption spectra of 9b, 9c and 9e exhibit a clear red shift of about 13 nm relative to 9a, while compound 10 (tetramethyl-substituted on the cyclophane) shows a significant blue shift (about 17 nm) in absorption bands in comparison to 9a.
| Compound | λ max/nm | Φ PL (%)a | |
|---|---|---|---|
| Absorption | Emission | ||
| a The PL efficiency (ΦPL) of the diethynyldithia[3.3]-paracyclophanes in CH2Cl2 solutions was measured by using quinine sulfate (ca. 1 × 10−5 M solution in 0.1 M H2SO4, assuming a ΦPL of 0.55) as a standard. | |||
| 9a | 240, 271 | 398 | 12 |
| 9b | 240, 284 | 402 | 9 |
| 9c | 240, 283 | 402 | 20 |
| 9d | 240, 274 | 400 | 22 |
| 9e | 241, 284 | 399 | 18 |
| 10 | 241, 254 | 400 | 5 |
| 11 | 240, 274 | 399 | 15 |
A comparison (Table 1) of the photoluminescence (PL) spectra of compounds 9a–e, 10 and 11 in CH2Cl2 (ca. 1 × 10−5 M) is illustrated in Fig. 3. The PL intensity of 9d (ΦPL = 22%) is significantly higher than that of 9a (ΦPL = 12%). An unexpected observation is that the ΦPL of 9c (dicyano-substituted, 20%) is considerably larger than that of 9a, while the PL of 9b (ΦPL = 9%) and 10 (ΦPL = 5%) are essentially decreased; the electron-donating dimethoxy substituents seem to behave as a quencher,11a,12,13 and the additional two methyls substituted in the pseudo-gem position appear to have a significant affect on the electronic properties of 10 (see also the absorption spectra and electronic structure, in which the dialkyne C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C unit is involved in the HOMO orbits, in contrast to the relevant dithia[3.3]paracyclophane disubstituted in the pseudo-meta position).
C unit is involved in the HOMO orbits, in contrast to the relevant dithia[3.3]paracyclophane disubstituted in the pseudo-meta position).
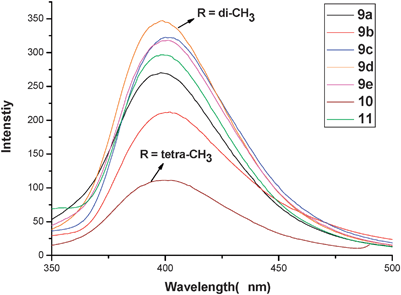 | ||
| Fig. 3 PL spectra of complexes 9a–e, 10 and 11 measured from solutions (ca. 1 × 10−5 M) in CH2Cl2 at room temperature. | ||
Furthermore, DFT computations at the B3LYP/6-31+G(d,p) level reveal that the electron density distribution in the relevant HOMO and LUMO are significantly different after the introduction of various substituents (Fig. 4). The above result is a good indication that the introduction of different substituents causes significant changes to the electronic and optical properties of the [3.3]paracyclophane core system via a transannular substitution effect.
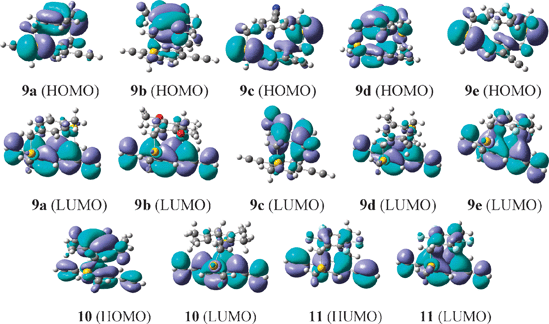 | ||
| Fig. 4 Optimized ground state structures, and the HOMO (H) and LUMO (L) electron densities of 9a–e, 10 and 11, as calculated by DFT (B3LYP/6-31+G(d,p)). | ||
Conclusions
In conclusion, a series of substituted dithia[3.3]paracyclophane dialkyne building blocks have been synthesized. UV-vis and photoluminescence studies and DFT calculations show that the electronic properties of these dialkynes can be tuned through a transannular substitution effect after the introduction of different substituents into the dithia[3.3]paracyclophane unit. These novel dialkyne building blocks should be valuable for extended carbon-rich π-systems,14 since their ethynyl functions can easily be connected by various reactions.Experimental
General considerations
All manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques, unless otherwise stated. 1H, 13C and 19F NMR spectra were collected on an American Varian Mercury Plus 400 (400 MHz) or 600 (600 MHz) spectrometer. 1H and 13C NMR chemical shifts are relative to TMS, and 19F NMR chemical shifts are relative to C6F6. High resolution ESI mass spectra were obtained on Bruker Apex IV FTMS spectrometers. UV-vis spectra were obtained on a S-3100 PDA UV-vis spectrometer (Scinco Co., Ltd.). Fluorescence spectra were acquired on a Fluoromax-P luminescence spectrometer (Horiba Jobin Yvon Inc.).2,5-Dibromo-1,4-bis(mercaptomethyl)benzene (1), 152e16 and 4b17 were prepared by standard literature methods. 2d, 4a were purchased from Tokyo Chemical Industry Co., Ltd. All other reagents were commercially available.
General procedure for the preparation of 3a–e, 5a and 5b
The compounds 3a–c were prepared according to a modified literature procedure.11cGeneral procedure: A solution with equimolar amounts of the dithiol and the dibromide in de-gassed THF (500 mL) was added dropwise under N2 over 12 h to a refluxing solution of K2CO3 (5 equiv.) in EtOH (1.2 L). After an additional 2 h at the reflux temperature, the mixture was cooled and the solvent removed. The resulting residue was treated with CH2Cl2 (300 mL) and water (300 mL). The organic phase was separated and the aqueous extracted three times with CH2Cl2. The combined organic layers were dried over Na2SO4, the solvent removed and the resulting solid chromatographed on silica gel using CH2Cl2/petroleum ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as the eluent. The product was further purified by recrystallization from toluene.
:1, v/v) as the eluent. The product was further purified by recrystallization from toluene.
General procedure for the preparation of 6a–e
A mixture of the appropriate dibromo-substituted paracyclophane 3a–e, Pd(PPh3)4 (10 mol%) and CuI (15 mol%) in toluene/Et3N (v/v = 5:2) was stirred at room temperature for 2 h under N2; then (trimethylsilyl)acetylene (in the molecular ratio 1:10) was added to the stirred solution. The mixture was stirred at 100 °C for 2 d under an N2 atmosphere. The precipitated ammonium salts were filtered off and the filtrate was evaporated under vacuum. The residue was subjected to column chromatography on silica gel using CH2Cl2/petroleum ether (1:1, v/v) as the eluent.
C), 122.82, 128.22, 129.15 133.47, 135.18, 137.80 (Ar). Anal. calc. for C26H32S2Si2: C, 67.18; H, 6.94. Found: C, 67.21; H, 6.62.
C), 112.67, 122.76, 123.89, 132.72, 137.89, 150.18 (Ar). Anal. calc. for C28H36O2S2Si2: C, 64.07; H, 6.91. Found: C, 64.17; H, 7.07.
C), 114.25 (CN), 115.83, 116.75, 123.49, 133.65, 137.41, 140.18 (Ar). Anal. calc. for C28H30N2S2Si2: C, 65.32; H, 5.87; N, 5.44. Found: C, 65.40; H, 5.43; N, 5.45.
C), 122.95, 131.73, 132.17, 132.84, 133.93, 137.51 (Ar). Anal. calc. for C28H36S2Si2: C, 68.23; H, 7.36. Found: C, 68.38; H, 7.79.
C), 116.91 (d, J = 29.85 Hz, Ar), 123.25, 124.39 (d, J = 25.35 Hz, Ar), 132.94, 137.64, 154.94, 156.53 (Ar). 19F NMR (376 MHz, CDCl3): δ −122.34. Anal. calc. for C26H30F2S2Si2: C, 62.35; H, 6.04. Found: C, 62.36; H, 6.17.
C), 121.55, 131.32, 132.48, 133.44, 134.16, 137.72 (Ar). Anal. calc. for C30H40S2Si2: C, 69.17; H, 7.74. Found: C, 69.14; H, 7.89.
:1) was added Pd(PPh3)2Cl2 (340 mg, 10 mol%) and CuI (91 mg, 10 mol%) under N2; the mixture was then refluxed for 48 h. The cooled reaction mixture was filtered and the filtrate evaporated under vacuum. The residue was subjected to column chromatography on silica gel using ethyl acetate/petroleum ether (1:2, v/v) as the eluent to give 0.76 g of 8 as a white solid (yield: 31%). 1H NMR (400 MHz, DMSO): δ 1.52 (s, 12H, CH3), 3.83 (q, J = 15.2 Hz, 4H, CH2), 3.99 (d, J = 14.8 Hz, 2H, CH2), 4.15 (d, J = 14.8 Hz, 2H, CH2), 5.49 (s, 2H, OH), 7.31 (s, 2H, Ar). 13C NMR (100 MHz, CDCl3): δ 23.81 (CH3), 31.48, 33.91 (CH2), 63.87 (C–OH), 78.06, 102.29 (CC), 121.97, 131.97, 137.27 (Ar); no other signals for the aromatic ring carbons were detected due to poor solubility. Anal. calc. for C26H24F4O2S2: C, 61.40; H, 4.76. Found: C, 61.16; H, 4.40.
General procedure for the preparation of 9a–e
To a stirred solution of bis(trimethylsilylethynyl)[3.3]dithia-paracyclophane (6a–e) in THF/MeOH (v/v = 1:1) was added 2.5 equiv. of KOH under N2; the reaction mixture was then stirred at room temperature for 2 h. The reaction mixture was diluted with dichloromethane and washed with brine. The organic layer was then dried over NaSO4 and the solvent removed in vacuo. The crude product was purified by chromatography on silica gel eluted with CH2Cl2/petroleum ether (1:1, v/v).
CH), 3.57 (d, J(HH) = 15.6 Hz, 2H, CH2), 3.78 (d, J(HH) = 14.4 Hz, 2H, CH2), 3.85 (d, J(HH) = 14.4 Hz, 2H, CH2), 4.28 (d, J(HH) = 15 Hz, 2H, CH2), 7.01 (d, J(HH) = 7.8 Hz, 2H, Ar), 7.05 (d, J(HH) = 7.8 Hz, 2H, Ar), 7.12 (s, 2H, Ar). 13C NMR (150 MHz, CDCl3): δ 34.87 (CH2), 37.79 (CH2), 81.94, 83.12 (CC), 122.15, 128.34, 129.26, 134.17, 135.25, 138.15 (Ar). MS(m/z): 320.8 (M+). HRMS (ESI): m/z [M + H]+ calc. for C20H17S2: 321.07662. Found: 321.07673. Anal. calc. for C20H16S2: C, 74.96; H, 5.03. Found: C, 75.21; H, 5.03.
CH), 3.50 (d, J = 16.8 Hz, 2H, SCH2), 3.64 (d, J = 16 Hz, 2H, SCH2), 3.89 (s, 6H, OCH3), 4.06 (q, J = 14.4 Hz, 4H, SCH2), 6.67 (s, 2H, Ar), 7.27 (s, 2H, Ar). 13C NMR (100 MHz, CDCl3): δ 31.99, 35.17 (SCH2), 55.38 (OCH3), 82.13, 83.94 (CC), 112.74, 122.01, 123.85, 133.21, 138.16, 150.16 (Ar). HRMS (ESI): m/z [M + H]+ calc. for C22H21O2S2: 381.09775. Found: 381.09759. Anal. calc. for C22H20O2S2: C, 69.44; H, 5.30. Found: C, 69.54; H, 5.23.
CH), 3.74 (d, J = 14.4 Hz, 2H, CH2), 3.86 (d, J = 16 Hz, 2H, CH2), 4.08 (d, J = 14.4 Hz, 2H, CH2), 4.17 (d, J = 14.8 Hz, 2H, CH2), 7.36 (s, 2H, Ar), 7.67 (s, 2H, Ar). 13C NMR (100 MHz, CDCl3): δ 35.13, 35.33 (CH2), 80.40, 85.34 (CC), 115.83 (CN), 116.78, 122.85, 133.81, 134.07 137.94, 140.33 (Ar). HRMS (ESI): m/z [M + H]+ calc. for C22H15N2S2: 371.06712. Found: 371.06711. Anal. calc. for C22H14N2S2: C, 71.32; H, 3.81; N, 7.56. Found: C, 71.66; H, 3.47; N, 7.42.
CH), 3.59 (d, J = 15.6 Hz, 2H, CH2), 3.74 (q, J = 15.6 Hz, 4H, CH2), 4.18 (d, J = 15.6 Hz, 2H, CH2), 6.80 (s, 2H, Ar), 7.38 (s, 2H, Ar). 13C NMR (150 MHz, CDCl3): δ 18.50 (CH3), 34.24, 35.90 (CH2), 81.57, 83.05 (CC), 122.21, 131.77, 132.51, 132.88, 133.96, 137.90 (Ar). HRMS (ESI): m/z [M + H]+ calc. for C22H21S2: 349.10792. Found: 349.10833. Anal. calc. for C22H20S2: C, 75.82; H, 5.78. Found: C, 75.63; H, 5.96.
CH), 3.54 (d, J = 16 Hz, 2H, SCH2), 3.68 (d, J = 14.4 Hz, 2H, SCH2), 3.91 (d, J = 14.4 Hz, 2H, SCH2), 4.14 (d, J = 14.8 Hz, 2H, SCH2), 6.88 (t, J = 8.8 Hz, 2H, Ar), 7.34 (s, 2H, Ar). 13C NMR (100 MHz, CDCl3): δ 30.56, 35.32 (SCH2), 81.23, 84.21 (CC), 117.08 (d, J = 29.3 Hz, Ar), 122.69, 124.55 (t, J = 12.2 Hz, Ar), 133.57, 138.06, 154.60, 157.00 (Ar). 19F NMR (376 MHz, CDCl3): δ −122.41. HRMS (ESI): m/z [M + H]+ calc. for C20H15F2S2: 357.05777. Found: 357.05793. Anal. calc. for C20H14F2S2: C, 67.39; H, 3.96. Found: C, 67.12; H, 3.64.
CH), 3.61 (d, J = 15.6 Hz, 2H, CH2), 3.68 (d, J = 15 Hz, 2H, CH2), 4.33 (d, J = 15.6 Hz, 2H, CH2), 4.38 (d, J = 15.6 Hz, 2H, CH2), 7.50 (s, 2H, Ar). 13C NMR (150 MHz, CDCl3): δ 17.10, 17.73 (CH3), 32.11, 33.69 (CH2), 81.99, 83.60 (CC), 120.84, 131.42, 132.77, 133.41, 134.10, 138.12 (Ar). HRMS (ESI): m/z [M + H]+ calc. for C24H25S2: 377.13922. Found: 377.13963. Anal. calc. for C24H24S2: C, 76.55; H, 6.42. Found: C, 76.50; H, 6.15.
:1) to yield 0.37 g of 11 as a white solid, yield: 64%. 1H NMR (600 MHz, CDCl3): δ 3.47 (s, 2H, CH), 3.69 (q, J = 15.6 Hz, 4H, SCH2), 4.04 (d, J = 15 Hz, 2H, SCH2), 4.36 (d, J = 15.6 Hz, 2H, SCH2), 7.55 (s, 2H, Ar). 13C NMR (150 MHz, CDCl3): δ 24.34, 34.58 (SCH2), 80.60, 84.34 (CC), 114.74, 122.24, 133.50, 137.95, 143.09, 144.11, 144.82, 145.78 (Ar). 19F NMR (376 MHz, CDCl3): δ −142.18. HRMS (ESI): m/z [M + H]+ calc. for C20H13F4S2: 393.03893. Found: 393.03932. Anal. calc. for C20H12F4S2:C, 61.21; H, 3.08. Found: C, 61.30; H, 3.33.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (nos. 2072039, 20931006, 21072070) and the Program for Changjiang Scholars and Innovative Research Team in University (no. IRT0953).References
- (a) S. Dahmen and S. Bräse, J. Am. Chem. Soc., 2002, 124, 5940 CrossRef CAS; (b) B. Jiang, Y. Lei and X.-L. Zhao, J. Org. Chem., 2008, 73, 7833 CrossRef CAS; (c) J. Ruan, L. Shearer, J. Mo, J. Bacsa, A. Zanotti-Gerosa, F. Hancock, X. Wu and J. Xiao, Org. Biomol. Chem., 2009, 7, 3236 RSC.
- (a) G. C. Bazan, J. Org. Chem., 2007, 72, 8615 CrossRef CAS; (b) E. Elacqua, D.-K. Bucar, Y. Skvortsova, J. Baltrusaitis, M. L. Geng and L. R. MacGillivray, Org. Lett., 2009, 11, 5106 CrossRef CAS.
- (a) Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2006, 45, 6430 CrossRef CAS; (b) H. Hopf, Angew. Chem., Int. Ed., 2008, 47, 9808 CrossRef CAS.
- A. Kahnt, D. M. Guldi, A. Escosura, M. V. Martínez-Díazb and T. Torres, J. Mater. Chem., 2008, 18, 77 RSC.
- (a) S. Clément, S. M. Aly, D. Fortin, L. Guyard, M. Knorr, A. S. Abd-El-Aziz and P. D. Harvey, Inorg. Chem., 2008, 47, 10816 CrossRef CAS; (b) H. Hopf, S. Sankararaman, I. Dix, P. G. Jones, H. G. Alt and A. Licht, Eur. J. Inorg. Chem., 2002, 123 CrossRef CAS; (c) P. J. Ball, T. R. Shtoyko, J. A. K. Bauer, W. J. Oldham and W. B. Connick, Inorg. Chem., 2004, 43, 622 CrossRef CAS.
- (a) D. Bléger, D. Kreher, F. Mathevet, A. Attias, I. Arfaoui, G. Metgé, L. Douillard, C. Fiorini-Debuisschert and F. Charra, Angew. Chem., Int. Ed., 2008, 47, 8412 CrossRef CAS; (b) L. Valentini, A. Taticchi, A. Marrocchib and J. M. Kennya, J. Mater. Chem., 2008, 18, 484 RSC.
- (a) L. Valentini, A. Marrocchi, M. Seri, F. Mengoni, F. Meloni, A. Taticchi and J. M. Kenny, Thin Solid Films, 2008, 516, 7193 CrossRef CAS; (b) F. Silvestri and A. Marrocchi, Int. J. Mol. Sci., 2010, 11, 1471 Search PubMed.
- (a) R. Cleiter and H. Hopf, Modern Cyclophane Chemistry, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (b) F. Vögtle, Cyclophane Chemistry, Wiley & Sons, New York, 1993 Search PubMed.
- (a) S. Clément, L. Guyard, M. Knorr, S. Dilsky, C. Strohmann and M. Arroyo, J. Organomet. Chem., 2007, 692, 839 CrossRef CAS; (b) A. Taticchi, A. Marrocchi, L. Minuti, S. Lundi and E. Gacs-Baitz, Heterocycles, 2006, 68, 1249 CrossRef CAS; (c) L. Minuti, A. Taticchi, A. Marrocchi, S. Landia and E. Gacs-Baitz, Tetrahedron Lett., 2005, 46, 5735 CrossRef CAS; (d) A. Marrocchi, A. Taticchi and S. Landi, Synthesis, 2008, 1045 Search PubMed; (e) L. Valentini, F. Mengoni, A. Taticchi, A. Marrocchi, S. Landi, L. Minutib and J. M. Kenny, New J. Chem., 2006, 30, 939 RSC; (f) L. Valentini, F. Mengoni, A. Taticchi, A. Marrocchi and J. M. Kenny, J. Mater. Chem., 2006, 16, 1622 RSC; (g) L. Valentini, A. Taticchi, A. Marrocchi and J. M. Kenny, J. Mater. Chem., 2008, 18, 484 RSC; (h) L. Valentini, F. Mengoni, J. M. Kenny, A. Marrocchi and A. Taticchi, Small, 2007, 3, 1200 CrossRef CAS; (i) L. Bgondarenko, I. Dix, H. Hinrichs and H. Hopf, Synthesis, 2004, 2751 Search PubMed; (j) H. Hopf and I. Dix, Synlett, 2006, 1146 Search PubMed; (k) H. Hopf, Tetrahedron, 2008, 64, 11504 CrossRef CAS , and references therein.
- See, for example: (a) Y. Morisaki, T. Murakami and Y. Chujo, Macromolecules, 2008, 41, 5960 CrossRef CAS; (b) G. J. Rowlands, Org. Biomol. Chem., 2008, 6, 1527 RSC.
- (a) W. Wang, J. Xu and Y.-H. Lai, Org. Lett., 2003, 5, 2765 CrossRef CAS; (b) W. Wang, J. Xu, Y.-H. Lai and F. Wang, Macromolecules, 2004, 37, 3546 CrossRef CAS; (c) W. Wang, J. Xu, Z. Sun, X. Zhang, Y. Lu and Y.-H. Lai, Macromolecules, 2006, 39, 7277 CrossRef CAS.
- V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924 CrossRef CAS.
- (a) G. J. Kavarnos and J. T. Turro, Chem. Rev., 1986, 86, 401 CrossRef CAS; (b) J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- Carbon-Rich Compounds II, Topics in Current Chemistry, ed. A. de Meijere, Springer, Berlin, 1999, vol. 201 Search PubMed.
- K.-Y. Kay and Y. G. Baek, Chem. Ber., 1997, 130, 581 CrossRef CAS.
- X.-S. Huang and F.-L. Qing, J. Fluorine Chem., 2008, 129, 1076 CrossRef CAS.
- F. C. Krebs and T. Jensen, J. Fluorine Chem., 2003, 120, 77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of bond distances and angles, X-ray crystal data for complex 5b, and UV-vis absorption spectra of 9a–e, 10 and 11. See DOI: 10.1039/c0nj00553c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |