Neodymium borohydride complexes supported by diamino-bis(phenoxide) ligands: diversity of synthetic and structural chemistry, and catalytic activity in ring-opening polymerization of cyclic esters†
Mikhail A.
Sinenkov
a,
Georgy K.
Fukin
a,
Anton V.
Cherkasov
a,
Noureddine
Ajellal
b,
Thierry
Roisnel
b,
Francesca M.
Kerton
c,
Jean-François
Carpentier
*b and
Alexander A.
Trifonov
*a
aG.A. Razuvaev Institute of Organometallic Chemistry of Russian Academy of Sciences Tropinina 49, GSP-445, 603950 Nizhny Novgorod, Russia. E-mail: trif@iomc.ras.ru; Fax: +7 8314621497
bOrganometallics and Catalysis, Institute of Chemistry, UMR 6226 CNRS-University of Rennes 1, 35042 Rennes Cedex, France. E-mail: jcarpent@univ-rennes1.fr; Fax: +33 223236939
cDepartment of Chemistry, Memorial University of Newfoundland, St. John’s, NL A1B 3X7, Canada. Fax: +1 709-7373702
First published on 19th October 2010
Abstract
New heterobimetallic borohydrido neodymium complexes {[OONN]1Nd(BH4)(μ-BH4)Li(THF)}2 (1) and [OONN]3Nd(BH4)(μ-BH4)Li(THF)2 (3) supported by diamino-bis(phenoxide) ligands ([OONN]1 = {CH2N(Me)CH2-3,5-Me,t-Bu-C6H2O}2; [OONN]3 = C5H4NCH2N{CH2-3,5-Me,t-Bu-C6H2O}2) were synthesized by the reactions of Nd(BH4)3(THF)2 with equimolar amounts of dilithium derivatives of diamino-bis(phenol)s Li2[OONN]n and isolated in high yields. In the case of Li2[OONN]2 ([OONN]2 = Me2NCH2CH2N{CH2-3,5-t-Bu-C6H2O}2), the same synthetic procedure afforded the heterobimetallic bis(phenoxide) complex Li{Nd[OONN2]2} (2). The structures of complexes 1–3 were established by X-ray diffraction studies. Compounds 1–3 act as single-site initiators for the ring-opening polymerization (ROP) of racemic lactide and racemic β-butyrolactone under mild conditions (20 °C), providing atactic polymers with controlled molecular weights and relatively narrow polydispersities (Mw/Mn = 1.07–1.82). While 1 and 3 initiate polymerizationvia their borohydride groups, ROP with 2 proceeds viainsertion into the Nd–O(ligand) bond.
Introduction
Although metallocene-type lanthanide borohydrides have been known for the past three decades,1 they have only recently been introduced in catalysis.2 In particular, the borohydride ligand turned out to be a promising active group for the synthesis of well-defined heteroleptic lanthanide complexes of the type [L]3−xLn(BH4)x (where L is a spectator mono- or dianionic ligand) that act as single-site initiators for the polymerization of a variety of monomers. For instance, metallocene-type lanthanide borohydrides were reported to initiate the polymerization of methyl methacrylate2e and the ring-opening polymerization (ROP) of ε-caprolactone,2b providing the corresponding polymers with a good degree of control over molecular weights. Also, when associated with alkyllithium or alkylmagnesium reagents, lanthanide borohydrides afford two-component catalytic systems for oligo/polymerizations of α-olefins, dienes and styrenes.2c,d,f,g,hAt the same time, the development of new ancillary (mainly N- and O-containing) ligand systems with various topologies, and steric and electronic properties is currently attracting growing attention. This offers an efficient approach to overcome some limitations of the cyclopentadienyl paradigm in tuning the metal atom coordination environment and eventually increasing the catalyst performance.3 Thus, diamide-diamine,4diamino-bis(phenoxide)5 and guanidinate6–8 ligands have allowed the synthesis of lanthanide borohydride complexes that have demonstrated high catalytic activity in controlled polymerizations of methyl methacrylate,4,7,8lactide5,6 and ε-caprolactone.4,5
Linked dianionic bis(phenoxide) ancillary ligands containing backbones of different length, structure, donor atom sets and possessing various denticity have a number of advantages: they are usually easy to prepare, they often prevent ligand redistribution reactions, and provide the metal center with a rigid framework enabling stereospecific transformations.9 These ligands have been successfully employed in organolanthanide chemistry to synthesize diverse types of complexes.10–27 In particular, alkoxy, amido and alkyl lanthanide species supported by tri- and tetradentate bis(phenoxide) ligands proved to be efficient initiators/catalysts for ROP of cyclic esters.10–19,21,24,26,27
Recently, we have reported on novel lanthanide borohydride6 and alkoxide28 complexes supported by bulky guanidinate ligands that promote the “controlled-living” and “immortal” ROP of racemic lactide and racemic β-butyrolactone. In the series of tested compounds, the neodymium derivatives demonstrated high initiation efficiency, good control over polymer molecular weight and polydispersity, and the ability to convert larger loadings of monomer.
In light of the above points, we focused our attention on the synthesis of related neodymium borohydride derivatives for evaluation in ROP of cyclic esters. Herein, we report on the synthesis and structures of neodymium borohydride complexes coordinated by diamino-bis(phenoxide) ligands and their catalytic activity in ROP of racemic lactide and racemic β-butyrolactone. Since the ancillary ligand is known to exert a crucial influence on the polymerization performance, diamino-bis(phenoxide) ligands of varying natures, including structure of the backbone and substituents on the phenyl rings, were investigated.
Results and discussion
1 Synthesis and structural characterization of complexes
In order to explore the influence of ancillary ligands (structure, nature and length of the linker between two phenol moieties; substituents on the phenyl rings) on the synthetic outcome and catalytic performance of the corresponding neodymium borohydrido species, three different diamino-bis(phenol)s H2[OONN]n possessing similar denticity were used (Chart 1).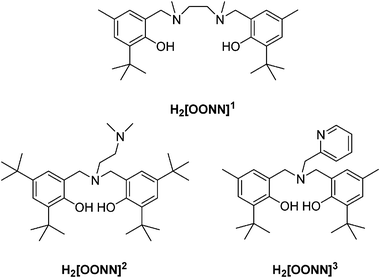 | ||
| Chart 1 | ||
Mountford and co-workers have described the synthesis of the first lanthanide borohydride complexes supported by pyridine containing diamino-bis(phenoxide) ligand,5 which were obtained by the reactions of Ln(BH4)3(THF)329 with disodium derivatives of the parent diamino-bis(phenol). In our previous studies, we found that the reactions of bis(guanidinate) chloro derivatives [{(Me3Si)2NC(Ni-Pr)2}2LnCl]2 (Ln = Nd, Sm) with NaBH4 are more complicated than those of cyclopentadienyl derivatives, and they were found to be inefficient as a synthetic pathway to bis(guanidinate) borohydrides.30 In fact, reactions of sodium guanidinates with Ln(BH4)3(THF)n in some cases occurred with fragmentation of the guanidine fragment or ligand redistribution,31 while the use of lithium derivatives cleanly afforded the borohydrido complexes [(Me3Si)2NC(N-R)2]2Ln(μ-BH4)2Li(THF)2 (R = Cy, iPr; Ln = Nd, Sm, Yb) in reasonable yields.6,7
Accordingly, in the present work, we focused on the metathesis reactions of Nd(BH4)3(THF)2 with dilithium diamino-bis(phenoxide)s (Li2[OONN]n). Lithiation of the diamino-bis(phenol)s was performed with two equivalents of n-BuLi in hexane at room temperature and the dilithium bis(phenoxide)s were used in situ for reaction with Nd(BH4)3(THF)2 either in toluene or THF solutions at 60 °C.
The reaction of Nd(BH4)3(THF)2 with an equimolar amount of Li2[OONN]1 in toluene afforded complex {[OONN]1Nd(BH4)(μ-BH4)Li(THF)}2 (1) (Scheme 1), which was isolated in 83% yield after recrystallization from hexane. Violet transparent crystals of 1 suitable for X-ray single crystal structure investigation were obtained by prolonged cooling of its toluene solution at −18 °C. Compound 1 crystallizes in P21/n space group; the unit cell contains two molecules of the complex and two solvate molecules of toluene. The molecular structure of 1 is depicted in Fig. 1; the crystal and structural refinement data, and selected bond lengths and angles for 1 are listed in Table 1 and Table 2, respectively.
 | ||
| Scheme 1 | ||
![ORTEP diagram (30% probability thermal ellipsoids) of {[OONN]1Nd(BH4)(μ-BH4)Li(THF)}2 (1) showing the non-hydrogen atom numbering scheme. Hydrogen atoms, methyl groups of t-Bu substituents and methylene groups of THF are omitted for clarity. Atoms with an “A” label are at equivalent position (−1 − x, −y, −1 − z).](/image/article/2011/NJ/c0nj00486c/c0nj00486c-f1.gif) | ||
| Fig. 1 ORTEP diagram (30% probability thermal ellipsoids) of {[OONN]1Nd(BH4)(μ-BH4)Li(THF)}2 (1) showing the non-hydrogen atom numbering scheme. Hydrogen atoms, methyl groups of t-Bu substituents and methylene groups of THF are omitted for clarity. Atoms with an “A” label are at equivalent position (−1 − x, −y, −1 − z). | ||
| 1·(C7H8) | 2·(C6H14) | 3 | |
|---|---|---|---|
| Distance/Å | |||
| Nd–B(1) | 2.808(2) | 2.747(3) | |
| Nd–B(2) | 2.667(2) | 2.671(3) | |
| Nd–H | 2.39(2)–2.61(2) | 2.43(3)–2.58(4) | |
| Nd–O(1) | 2.230(1) | 2.168(2) | |
| Nd–O(2) | 2.184(1) | 2.359(2) | |
| Nd–O(1B) | 2.298(3) | ||
| Nd–O(2B) | 2.258(3) | ||
| Nd–N(1) | 2.654(1) | 2.616(2) | |
| Nd–N(2) | 2.628(2) | 2.608(2) | |
| Nd–N(1B) | 2.620(4) | ||
| Nd–N(2B) | 2.740(4) | ||
| Li–B(1) | 2.520(4) | 2.541(5) | |
| Li–B(1A) | 2.690(4) | ||
| Li–H | 2.07(2)–2.20(2) | 2.10(3)–2.18(3) | |
| Li–O(1A) | 1.953(9) | ||
| Li–O(2A) | 2.024(9) | ||
| Li–N(1A) | 2.063(9) | ||
| Li–N(2A) | 2.17(1) | ||
| Angles (°) | |||
| B(1)–Nd–B(2) | 100.99(7) | 101.9(1) | |
| O(1)–Nd–O(2) | 164.53(4) | 150.70(6) | |
| O(1B)–Nd–O(2B) | 151.8(1) | ||
| N(1)–Nd–N(2) | 67.63(5) | 66.31(6) | |
| N(1B)–Nd–N(2B) | 66.6(1) | ||
| O(1A)–Li–O(2A) | 96.3(4) | ||
| N(1A)–Li–N(2A) | 86.8(3) | ||
| 1·(C7H8) | 2·(C6H14) | 3 | |
|---|---|---|---|
| Empirical formula | C71H124B4Li2N4Nd2O6 | C74H122LiN4NdO4 | C38H62B2LiN2NdO4 |
| Formula weight | 1475.34 | 1282.94 | 783.7 |
| Crystal size/mm | 0.16 × 0.12 × 0.07 | 0.35 × 0.28 × 0.17 | 0.46 × 0.31 × 0.24 |
| T/K | 100(2) | ||
| Space group | P21/n | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
| a/Å | 12.5495(4) | 15.0652(8) | 9.744(5) |
| b/Å | 24.3646(7) | 18.920(1) | 14.106(5) |
| c/Å | 13.2411(4) | 26.470(2) | 15.815(5) |
| α (°) | 90 | 90 | 73.379(5) |
| β (°) | 108.412(1) | 92.549(1) | 80.941(5) |
| γ (°) | 90 | 90 | 85.697(5) |
| V/Å3 | 3841.4(2) | 7537.3(7) | 2055.9(14) |
| Z | 2 | 4 | 2 |
| Calculated density/g cm−3 | 1.276 | 1.131 | 1.266 |
| μ/mm−1 | 1.385 | 0.734 | 1.300 |
| Absorption correction | SADABS | SADABS | SADABS |
| T min/Tmax | 0.8088/0.9093 | 0.7831/0.8853 | 0.555/0.732 |
| F(000) | 1544 | 2756 | 818 |
| 2θ/° | 52 | 48 | 50 |
| Unique reflections collected (Rint) | 7548 (0.0484) | 11789 (0.0847) | 8928 (0.0408) |
| R 1 (I > 2σ(I)) | 0.0292 | 0.0721 | 0.0283 |
| wR2 (all data) | 0.0662 | 0.2137 | 0.0708 |
| Parameters | 594 | 731 | 459 |
| Goodness-of-fit on F2 | 1.000 | 1.075 | 1.060 |
| Largest diff. hole and peak, e/Å3 | −0.428/1.297 | −3.743/3.095 | −1.226/1.487 |
The X-ray diffraction study revealed that 1 is a dimeric heterobimetallic complex in the solid state. Monoanionic borohydride groups are known to act in both μ-bridging and terminal coordination modes, which results in the formation of dimeric32 or monomeric organolanthanide complexes.33
Depending on the central atom size and its coordination environment, borohydride anions can act in monomeric complexes as bi-1,32a,33,34 and tridentate1,2b,2c,35 ligands. For dimeric complexes, μ2-η3:η2-bridging coordination has been described.32b,36 In compound 1, the coordination sphere of each neodymium atom contains one terminal and one μ-bridging BH4 groups. The μ-bridging BH4 groups connect one neodymium and two lithium ions, thus leading to the formation of a dimeric core. The terminal borohydrido group is bound to the neodymium atom in a η3-fashion, while the μ-bridging one coordinates to the lithium and neodymium atoms in a η2-mode. The Nd–H bond distances fall into the range 2.39(2)–2.61(2) Å, as usually observed for this kind of compound.2c,2d,6,7,36 As expected, the Nd–B bond distance for the terminal borohydrido groups (2.667(2) Å) is noticeably shorter compared to that for the bridging one (2.808(2) Å). Besides the two borohydrido groups, the coordination environment of each neodymium center is formed by two covalently bound oxygen atoms and two coordinatively bound nitrogen atoms, thus leading to a distorted octahedral geometry. The oxygen atoms are situated in the equatorial plane in trans positions (O(2)–Nd(1)–O(1) bond angle = 164.53(4)°). The Nd–O bond lengths in 1 (2.184(1) and 2.230(1) Å) are comparable to the values previously reported for related complexes.18,37 The methyl substituents of the nitrogen atoms adopt a trans-orientation relative to the N(1)–Nd–N(2) plane. The planes of the phenyl rings and the N(1)–N–N(2) plane are nearly orthogonal (the corresponding dihedral angles are 79.6° and 85.9°).
The IR spectrum of 1 in the region 2100–2500 cm−1 shows a set of four strong absorption bands, diagnostic of η3- and η2-bridging borohydride ligands.381H NMR spectroscopy was uninformative about the solution structures of complexes 1–3 due to the strong paramagnetism of the neodymium centers; only extremely broadened resonances were observed.
Attempts to synthesize the neodymium borohydrido complex supported by the related diamino-bis(phenoxide) ligand [OONN]2, which has an aliphatic linker of the same composition but of different structure (Chart 1), using an analogous synthetic approach, reaction conditions and workup as for 1, afforded an unexpected product. The reaction of Nd(BH4)3(THF)2 with an equimolar amount of Li2[OONN]2, in either toluene or THF at 60 °C, resulted in the formation of Li{Nd[OONN2]2} (2) (Scheme 2), which was isolated in 34% yield after recrystallization from hexane. Violet-blue transparent crystals of 2 suitable for X-ray diffraction studies were grown by slow concentration of a hexane solution at 20 °C.
 | ||
| Scheme 2 | ||
The X-ray crystal structure investigation revealed that compound 2 is a heterobimetallic complex that does not contain any borohydrido group (Fig. 2). This complex crystallizes as a solvate, 2·(C6H14), in the monoclinic space groupP21/n. The molecular structure of 2 contains one neodymium and one lithium atom and two diamino-bis(phenoxide) ligands that are linked to the metal centers in different fashions. One of the diamino-bis(phenoxide) fragments is coordinated only to the neodymium atom, while the second one forms M–O bonds with both the neodymium and lithium atoms. Both of the nitrogen atoms of each ligand are coordinated to either the neodymium or the lithium atoms, respectively. As a result, the geometries of the diamino-bis(phenoxide) ligands differ noticeably: the value of the dihedral angle between the phenyl ring planes in the ligand coordinated to the neodymium atom (140.5°) is much larger than the corresponding value observed for the ligand coordinated to Li and Nd (117.3°). The difference of the ion sizes of Nd3+ and Li+ influences strongly the distances between the oxygen atoms of the diamino-bis(phenoxide) ligands: 4.419 and 2.963 Å, respectively. The neodymium atom is coordinated by four oxygen and two nitrogen atoms and lies in a distorted octahedral environment. Expectedly, the Nd–O bond lengths for the terminal diamino-bis(phenoxide) ligand (2.258(3) and 2.298(3) Å) are somewhat shorter than for the bridging ones (2.322(3) and 2.325(3) Å). Despite the different coordination modes, the Nd−N bonds have rather similar values (2.620(4) and 2.740(4) Å).
![ORTEP diagram (30% probability thermal ellipsoids) of Li{Nd[OONN2]2} (2) showing the non-hydrogen atom numbering scheme. Hydrogen atoms and methyl groups of t-Bu substituents are omitted for clarity.](/image/article/2011/NJ/c0nj00486c/c0nj00486c-f2.gif) | ||
| Fig. 2 ORTEP diagram (30% probability thermal ellipsoids) of Li{Nd[OONN2]2} (2) showing the non-hydrogen atom numbering scheme. Hydrogen atoms and methyl groups of t-Bu substituents are omitted for clarity. | ||
The reaction of Nd(BH4)3(THF)2 with the dilithium derivative of the pyridyl-substituted ligand Li2[OONN]3, due to the limited solubility of the latter in toluene, was carried out in THF at 60 °C. This reaction allowed the synthesis of neodymium borohydrido complex 3 (Scheme 3), which was obtained in 74% yield after recrystallization from toluene at −20 °C.
 | ||
| Scheme 3 | ||
Single crystals of 3 suitable for X-ray diffraction study were obtained by prolonged cooling of its toluene solution at 5 °C. Compound 3 crystallizes in the P space group. The X-ray crystal structure investigation revealed that 3 is a monomeric heterobimetallic ate-complex containing one residual LiBH4 per (BH4)Nd[OONN]3 unit in which the neodymium and the lithium atoms are connected by one μ2-bridging BH4 and one μ2-phenoxide fragment (Fig. 3). The coordination sphere of the neodymium atom in 3 consists of the two oxygen and two nitrogen atoms of the diamino-bis(phenoxide) ligand and two borohydrido groups. The terminal borohydrido group is bound to neodymium in a η3-fashion, while the μ-bridging one is connected to the neodymium atom in an η3- and to the lithium atom in an η2-fashion. The Nd–H bond distances for the terminal group are in the range 2.46(4)–2.58(4) Å and the Nd–B distance is 2.671(3) Å. The analogous parameters for the μ-bridging group are somewhat longer (Nd–H, 2.43(3)–2.48(3) Å; Nd–B, 2.747(3) Å). The diamino-bis(phenoxide) ligand in 3 is bound to the neodymium atom by one short terminal Nd–O bond (2.168(2) Å) and by one longer bond (2.359(2) Å) involving the oxygen atom μ-bridging the neodymium and lithium atoms. The value of the dihedral angle between the planes of the phenyl rings is 123.9°. Similarly to complex 1, the IR spectrum of 3 contains in the region 2100–2500 cm−1 four strong absorptions indicative of η3- and η2-coordinated borohydrido groups.32b,33a
![ORTEP diagram (30% probability thermal ellipsoids) of [OONN]3Nd(BH4)(μ-BH4) Li(THF)2 (3) showing the non-hydrogen atom numbering scheme. Hydrogen atoms, methyl groups of t-Bu substituents and methylene groups of THF are omitted for clarity.](/image/article/2011/NJ/c0nj00486c/c0nj00486c-f3.gif) | ||
| Fig. 3 ORTEP diagram (30% probability thermal ellipsoids) of [OONN]3Nd(BH4)(μ-BH4) Li(THF)2 (3) showing the non-hydrogen atom numbering scheme. Hydrogen atoms, methyl groups of t-Bu substituents and methylene groups of THF are omitted for clarity. | ||
2 Ring-opening polymerization of racemic lactide and β-butyrolactone
The prepared [diamino-bis(phenoxide)]-lanthanide complexes 1–3 have been evaluated in the ROP of racemic lactide (rac-LA) and racemic β-butyrolactone (rac-BBL) (Scheme 4). Representative results are summarized in Tables 3 and 4, respectively. | ||
| Scheme 4 | ||
| Entry | Complex | [LA]/[Ln] | Solvent | Time/hb | Conv. (%)c | M n,calc d (×103) | M n,exp e (×103) | M w/Mne |
|---|---|---|---|---|---|---|---|---|
| a All reactions performed with [rac-LA] = 1.0 M at 20 °C, unless otherwise stated; results are representative of at least duplicated experiments. b Reaction times were not necessarily optimized. c Isolated yields of PLA. d M n (in g mol−1) of PLA calculated from Mn,calc = 144.00 × ([LA]/[Ln]) × yield(LA). e Experimental (corrected; see Experimental section) Mn (in g mol−1) and Mw/Mn values determined by SEC in THFvs.polystyrene standards. | ||||||||
| 1 | 1 | 50 | THF | 12 | >99 | 7.2 | 6.8 | 1.15 |
| 2 | 1 | 100 | THF | 12 | >99 | 14.2 | 13.3 | 1.25 |
| 3 | 1 | 200 | THF | 12 | 95 | 27.4 | 22.8 | 1.21 |
| 4 | 1 | 100 | Tol | 12 | 45 | 6.5 | 15.0 | 1.25 |
| 5 | 2 | 100 | THF | 12 | >99 | 14.2 | 10.0 | 1.36 |
| 6 | 2 | 500 | THF | 12 | 72 | 51.8 | 38.4 | 1.41 |
| 7 | 2 | 100 | Tol | 12 | 34 | 4.9 | 5.2 | 1.82 |
| 8 | 3 | 100 | Tol | 12 | 71 | 10.0 | 4.0 | 1.44 |
| 9 | 3 | 100 | THF | 12 | >99 | 14.2 | 8.1 | 1.55 |
| 10 | 3 | 200 | THF | 2 | 91 | 26.2 | 10.5 | 1.64 |
| 11 | 3 | 500 | THF | 5 | 83 | 59.7 | 15.0 | 1.75 |
| Entry | Complex | [BBL]/[Ln] | Solvent | Time/hb | Conv. (%)c | M n,calc d (×103) | M n exp e (×103) | M w/Mne |
|---|---|---|---|---|---|---|---|---|
| a All reactions performed with [rac-BBL] = 3.0 M at 20 °C, unless otherwise stated; results are representative of at least duplicated experiments. b Reaction times were not necessarily optimized. c Isolated yields of PHB. d M n (in g mol−1) of PHB calculated from Mn,calc = 86 × ([BBL]/[Ln]) × yield(BBL). e Experimental Mn (in g mol−1) and Mw/Mn values determined by SEC in THFvs.polystyrene standards. | ||||||||
| 12 | 1 | 100 | Tol | 12 | 61 | 5.3 | 5.0 | 1.07 |
| 13 | 1 | 100 | THF | 12 | 45 | 3.9 | 2.5 | 1.10 |
| 14 | 2 | 100 | THF | 12 | 87 | 7.5 | 9.1 | 1.32 |
| 15 | 2 | 100 | Hex | 12 | 89 | 7.7 | 6.4 | 1.38 |
| 16 | 2 | 100 | Tol | 12 | 99 | 8.5 | 6.9 | 1.29 |
| 17 | 2 | 200 | Tol | 16 | 95 | 16.3 | 10.4 | 1.31 |
| 18 | 2 | 500 | Tol | 24 | 60 | 25.8 | 12.3 | 1.55 |
| 19 | 2 | 1000 | Tol | 48 | 28 | 24.1 | 15.3 | 1.41 |
| 20 | 3 | 100 | Tol | 12 | 24 | 2.1 | 1.9 | 1.10 |
| 21 | 3 | 100 | THF | 12 | 24 | 2.1 | 2.5 | 1.12 |
These compounds are all active towards both monomers under mild conditions, allowing conversion of up to 400 equiv. of rac-LA and up to 300 equiv. of rac-BBL at room temperature in either toluene, hexane or THF solutions at [rac-LA] = 1.0 mol L−1 and [rac-BBL] = 3.0 mol L−1. Kinetic monitoring revealed, however, that catalytic activities are affected by the nature of the solvent. With the three complexes investigated, ROP of rac-LA proceeded faster in THF than in toluene (Table 3). This is in agreement with the observations of Bonnet and Mountford on related diamino-bis(phenolate)-lanthanide borohydride complexes.5 The solvent effect was much less pronounced for ROP reactions of rac-BBL, for which sensibly similar completion times were observed for each individual compound (1–3) in THF, toluene or hexane (Table 4).
All the PLAs and PHBs produced showed atactic microstructures, as determined by NMR analysis.15a,27d A control experiment performed with L-lactide and compound 3 resulted in pure isotactic PLA, supporting the lack of base-promoted epimerization of L-lactide or PLA and arguing against an anionic polymerization mechanism being operative.41
Experiments aimed at investigating the degree of control of polymerizations were carried out. All the PLAs and PHBs obtained with complexes 1–3 showed unimodal SEC traces with relatively narrow molecular weight in the range Mw/Mn = 1.07–1.82. Narrower distributions were observed with compound 1 for the ROP of rac-LA (Mw/Mn = 1.15–1.25) and with compounds 1 and 3 for the ROP of rac-BBL (Mw/Mn = 1.07–1.12), indicative of a single-site character under these conditions. Compound 2, which does not have any borohydride group in contrast to 1 and 3 (vide infra), yielded polymers with broader polydispersities.
For the ROP of rac-LA promoted by borohydride complexes 1 and 3, the number-average molecular masses (Mn) values increased monotonically (although not perfectly linearly) with the monomer-to-metal ratio (compare entries 1/2, and 9/10/11). The 1H NMR spectra in CDCl3 of relatively low molecular weight samples of PLA produced with 1 and 3 were identical to those we obtained previously with related borohydride-lanthanide complexes:6 they showed the quartet characteristic of the CH(Me)OH terminal group at δ 4.33 ppm, which arises from hydrolysis of the metal-alkoxide bond in the active species, an observation consistent with a classical coordination/insertion mechanism with an initial ring-opening viaacyl-oxygen bond cleavage. Additional resonances were observed, which include a doublet of doublet at δ 5.50 ppm and two doublet/quartet at δ 4.15 and 4.45 ppm, that could correspond to CH2OH end-groups, as expected from the [HBH3] functionality acting as both an initiating group and a reducing agent.6 Yet, as for several other polyesters prepared from borohydride-lanthanide catalyst systems, the exact nature of all termini could not be unambiguously determined.4–6 Note, however, that resonances diagnostic for the bis(phenolate) were not observed in the 1H NMR spectra of the polymers prepared from 1 and 3, suggesting that the ligand did not act (or not significantly) as an initiating group in those polymerizations, in contrast to those mediated by 2 (vide infra).
An issue with complexes 1 and 3 was to assess if they act as mono- or di-initiators; i.e., if only one or both borohydride groups initiate a polymer chain. The corrected experimental number-average molecular masses (Mn) of PLAs produced with 1 were in most cases close to the theoretical ones, calculated on the assumption that a single HBH3 group initiates the polymerization, that is a single PLA chain is produced per metal center. On the other hand, for PLAs produced by 3, the experimental Mn values were approximately half of the one calculated on the aforementioned principle, suggesting that both borohydride groups may be active in this compound and/or that different initiation processes are operative.
An unexpected result of this study was the significant activity of complex 2 toward both rac-LA and rac-BBL at room temperature, despite the fact that this compound contains no borohydride group. Yet, ROP of cyclic esters initiated by alkali phenolate compounds is now well documented,39 and this suggests how the mixed Nd–Li compound 2 may be operative in the experiments discussed herein. The experimental Mn values of PLAs produced with 2 were somewhat lower than those calculated on the basis of one growing polymer chain per metal center (Table 3). Increasing the lactide loading from 100 to 200 equiv. led to an almost doubled polymer weight, but higher loading (500–1000 equiv.) resulted in Mn values much smaller than those expected. A similar trend was observed in the ROP of rac-BBL (Table 4). These observations are consistent with ROP reactions initiated by compound 2 also via a coordination/insertion mechanism, with significant transfer reactions (to the monomer) occurring at larger monomer loadings. As mentioned above, we assumed that initiation with compound 2 would proceed viainsertion into any of the M–O(phenoxide-ligand) bonds (M = Li or Nd). To corroborate this hypothesis, a relatively low molecular weight PHB sample produced with 2 (Table 2, entry 16) was analyzed by NMR spectroscopy. The 1H NMR spectrum in CDCl3 of a twice-reprecipitated sample showed, in addition to the resonances characteristic for the PHB main chain, small resonances assignable to terminus groups (Fig. 4). Among these, several signals, including the aromatic hydrogens at 6.85 and 7.2 ppm, the benzylic hydrogens at 3.6 ppm and the dimethylamino group at 2.32 ppm, were clearly diagnostic of the presence of a ligand moiety [OONN]2 at one end of the polymer chain.
 | ||
| Fig. 4 1H NMR spectrum (500 MHz, CDCl3, 20 °C) showing the diagnostic resonances (*) assigned to the terminal groups of the polymer chain in a PHB sample prepared by polymerization of rac-BBL in toluene with complex 2 (Table 2, entry 16). | ||
Conclusions
The reactions of Nd(BH4)3(THF)2 with equimolar amounts of dilithium derivatives of diamino-bis(phenol)s Li2[OONN]n possessing similar denticity turned out to be sensitive to the structure, nature and length of the linker between the two phenol moieties. These reactions can afford dimeric {[OONN]1Nd(BH4)(μ-BH4)Li(THF)}2 (1) and monomeric [OONN]3Nd(BH4)(μ-BH4)Li(THF)2 (3) heterobimetallic borohydrido or heterobimetallic bis(phenolate) Li{{Nd[OONN2]2} (2) complexes. Borohydrido compounds 1 and 3 are active for the ROP of lactide and the more challenging β-butyrolactone as well, under mild conditions. Overall, their catalytic behavior can be related to that of neodymium borohydride complexes supported by bulky guanidinate ligands.6 No significant influence of the ancillary ligands could be evidenced. The notable reactivity of compound 2 towards both LA and BBL, although the latter compound bears no borohydride group, has been rationalized by insertion of the monomer into M–O(phenolate) bonds. These results illustrate the possible competition and noticeable differences that may occur in the initiation phase of ROP processes mediated by such families of rare-earth borohydride-phenolate compounds.Experimental
General conditions
All experiments were performed in evacuated tubes, using standard Schlenk techniques or a glove box, with rigorous exclusion of traces of moisture and air. After drying over KOH, THF and diethyl ether were purified by distillation from sodium/benzophenone ketyl, and hexane and toluene were dried by distillation from sodium/triglyme and benzophenone ketyl prior to use. Diamino-bis(phenol)s39 and Nd(BH4)3(THF)229 were prepared according to literature procedures. Racemic lactide (Aldrich) was recrystallized twice from dry toluene and then sublimed under vacuum at 50 °C. Racemic β-butyrolactone (Aldrich) was freshly distilled from CaH2 under nitrogen and degassed thoroughly by freeze-thaw-vacuum cycles prior to use. All other commercially available chemicals were used after the appropriate purification.Instruments and measurements
IR spectra of complexes were recorded as Nujol mulls on a Bruker-Vertex 70 spectrophotometer. C and H elemental analyses were performed by the microanalytical laboratory of IOMC. Lanthanide metal analysis was carried out by complexometric titration. Size exclusion chromatography (SEC) of PLAs and PHBs was performed in THF at 20 °C using a Polymer Laboratories PL50 apparatus equipped with PLgel 5 μm MIXED-C 300 × 7.5 mm columns, and combined RI and Dual angle LS (PL-LS 45/90°) detectors. The number average molecular masses (Mn) and polydispersity index (Mw/Mn) of the polymers were calculated with reference to a universal calibration vs.polystyrene standards. Mn values of PLAs were corrected with a factor of 0.58 to account for the difference in hydrodynamic volumes between PS and PLA.12Mn values of PHBs are uncorrected. The microstructure of PLAs was determined by homodecoupling 1H NMR spectroscopy at 20 °C in CDCl3 with a Bruker AC-500 spectrometer. The microstructure of PHBs was determined by analyzing the carbonyl region of 13C{1H} NMR spectra at 20 °C in CDCl3 with a Bruker AC-500 spectrometer operating at 125 MHz.27dSynthesis of complexes
X-Ray crystallography
The X-ray data were collected on a SMART APEX diffractometer (graphite-monochromated, Mo-Kα-radiation, ω- and θ-scan technique, λ = 0.71073 Å). The structures were solved by direct methods and were refined on F2 using SHELXTL41 package. All non-hydrogen atoms were found from Fourier syntheses of electron density and were refined anisotropically. The hydrogen atoms of BH4-groups were found from Fourier syntheses of electron density and were refined isotropically. Other hydrogen atoms were placed in calculated positions and were refined in the riding model. SADABS42 was used to perform area-detector scaling and absorption corrections. The quality of diffraction data for complex 1 did not allow to refine the coordinates and anisotropic displacement parameters of carbon atoms in the solvate molecule of toluene which lies in the inversion center. Therefore, the geometry of this toluene molecule was fixed by DFIX and ISOR instructions. The coordinates of carbon atoms and their anisotropic displacement parameters were “frozen” in the final refinement. The same situation was in solvate molecule of hexane in 2, which was treated similarly. The details of crystallographic, collection and refinement data are shown in Table 2. CCDC reference numbers 769020–769022. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0nj00486cPolymerization
Acknowledgements
This work is supported by the Russian Fundation for Basic Research (Grants N 08-03-00391-a; 09-03-97034-r_povolzh'e_a), Program of the Presidium of the Russian Academy of Science (RAS), the GDRI “CH2D” between RAS Chemistry and Material Science Division and CNRS, and the Institut Universitaire de France (IUF).References
- T. J. Marks and G. W. Grynkewich, Inorg. Chem., 1976, 15, 1302–1307 CrossRef CAS.
- (a) I. Palard, A. Soum and S. M. Guillaume, Chem.–Eur. J., 2004, 10, 4054–4062 CrossRef; (b) D. Barbier-Baudry, O. Blacque, A. Hafid, A. Nyassi, H. Sitzmann and M. Visseaux, Eur. J. Inorg. Chem., 2000, 2333–2336 CrossRef CAS; (c) M. Visseaux, T. Chenal, P. Roussel and A. Mortreux, J. Organomet. Chem., 2006, 691, 86–92 CrossRef CAS; (d) N. Barros, M. Schappacher, P. Dessuge, L. Maron and S. M. Guillaume, Chem.–Eur. J., 2008, 14, 1881–1890 CrossRef CAS; (e) P. Zinck, A. Valente, A. Mortreux and M. Visseaux, Polymer, 2007, 48, 4609–4614 CrossRef CAS.
- (a) F. T. Edelmann, D. M. M. Freckmann and H. Schumann, Chem. Rev., 2002, 102, 1851–1896 CrossRef CAS; (b) W. E. Piers and D. J. H Emslie, Coord. Chem. Rev., 2002, 233–234, 131–155 CrossRef CAS; (c) P. Mountford and B. D. Ward, Chem. Commun., 2003, 1797–1803 RSC; (d) A. Trifonov, Russ. Chem. Rev., 2007, 76, 1051–1072 Search PubMed.
- F. Bonnet, A. C. Hillier, A. Collins, S. R. Dubberley and P. Mountford, Dalton Trans., 2005, 421–423 RSC.
- F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046–9055 CrossRef CAS.
- G. G. Skvortsov, M. V. Yakovenko, P. M. Castro, G. K. Fukin, A. V. Cherkasov, J.-F. Carpentier and A. A. Trifonov, Eur. J. Inorg. Chem., 2007, 3260–3267 CrossRef CAS.
- G. G. Skvortsov, M. V. Yakovenko, G. K. Fukin, E. V. Baranov, Yu. A. Kurskii and A. A. Trifonov, Russ. Chem. Bull., 2007, 56, 456–460 Search PubMed.
- F. Yuan and L. Xiong, J. Organomet. Chem., 2006, 691, 3377–3382 CrossRef CAS.
- H. Kawaguchi, J. Organomet. Chem., 2004, 689, 4228–4243 CrossRef CAS.
- C.-X. Cai, L. Toupet, C. W. Lehmann and J.-F. Carpentier, J. Organomet. Chem., 2003, 683, 131–136 CrossRef.
- C.-X. Cai, A. Amgoune, C. W. Lehmann and J.-F. Carpentier, Chem. Commun., 2004, 330–331 RSC.
- H. Ma and J. Okuda, Macromolecules, 2005, 38, 2665–2673 CrossRef CAS.
- Y. Yao, M. Ma, X. Xu, Y. Zhang, Q. Shen and W.-T. Wong, Organometallics, 2005, 24, 4014–4020 CrossRef CAS.
- H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef.
- (a) A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169–179 CrossRef CAS; (b) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Pure Appl. Chem., 2007, 79, 2013–2030 CrossRef CAS.
- H. Ma, T. P. Spaniol and J. Okuda, Dalton Trans., 2003, 4770–4780 RSC.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- X. Xu, Z. Zhang, Y. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 9379–9288 CrossRef CAS.
- H. Zhou, H. Guo, Y. Yao, L. Zhou. H. Sun and H. Sheng, Inorg. Chem., 2007, 46, 958–964 CrossRef CAS.
- (a) M. Konkol, T. P. Spaniol, M. Kondracka and J. Okuda, Dalton Trans., 2007, 4095–4102 RSC; (b) M. Konkol, M. Kondracka, P. Voth, T. P. Spaniol and J. Okuda, Organometallics, 2008, 27, 3774–3784 CrossRef CAS.
- (a) H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32–35 RSC; (b) L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273–275 RSC.
- R. Qi, B. Liu, X. Xu, Z. Yang, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2008, 5016–5024 RSC.
- (a) N. Ikpo, S. M. Butt, K. L. Collins and F. M. Kerton, Organometallics, 2009, 28, 837–842 CrossRef CAS; (b) C. E. Willans, M. A. Sinenkov, G. K. Fukin, K. Sheridan, J. M. Lynam, A. A. Trifonov and F. M. Kerton, Dalton Trans., 2008, 3592–3598 RSC; (c) F. M. Kerton, A. C. Whitwood and C. E. Willans, Dalton Trans., 2004, 2237–2244 RSC.
- Z. Zhang, X. Xu, W. Li, Y. Yao, Y. Zhang, Q. Shen and Y. Luo, Inorg. Chem., 2009, 48, 5715–5724 CrossRef CAS.
- H. Yao, Y. Zhang, H. Sun and Q. Shen, Eur. J. Inorg. Chem., 2009, 1920–1925 CrossRef CAS.
- (a) J. W. Kramer, D. S. Treiter, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS; (b) J.-F. Carpentier, Angew. Chem., Int. Ed., 2010, 49, 2662–2663 CAS.
- (a) P. M. Castro, G. Zhao, A. Amgoune, C. M. Thomas and J.-F. Carpentier, Chem. Commun., 2006, 4509–4511 RSC; (b) A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS; (c) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS; (d) N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS; (e) N. Ajellal, C. M. Thomas and J.-F. Carpentier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3177–3189 CrossRef CAS; (f) E. Grunova, E. Kirillov, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2010, 39, 6739 RSC.
- N. Ajellal, D. M. Lyubov, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, C. M. Thomas, J.-F. Carpentier and A. A. Trifonov, Chem.–Eur. J., 2008, 14, 5440–5448 CrossRef CAS.
- U. Mirsaidov, I. Shaimuradov and M. Hikmatov, Zh. Neorg. Khim. (Rus. J. Inorg. Chem.), 1986, 31, 1321–1326 Search PubMed.
- A. A. Trifonov, G. G. Skvortsov, D. M. Lyubov, G. K. Fukin, E. A. Fedorova and M. N. Bochkarev, Russ. Chem. Bull., 2005, 54, 2511–2518 Search PubMed.
- (a) G. G. Skvortsov, D. M. Lyubov, M. V. Yakovenko, G. K. Fukin, A. V. Cherkasov and A. A. Trifonov, Izv. AN, Ser. Khim., 2009, 1098–1103 Search PubMed; (b) G. G. Skvortsov, M. V. Yakovenko, G. K. Fukin, A. V. Cherkasov and A. A. Trifonov, Russ. Chem. Bull., 2007, 56, 1742–1748 Search PubMed.
- (a) Y. K. Gun'ko, B. M. Bulychev, G. L. Soloveichik and V. K. Belsky, J. Organomet. Chem., 1992, 424, 289–300 CrossRef; (b) E. B. Lobkovsky, Y. K. Gun'ko, B. M. Bulychev, V. K. Belsky, G. L. Soloveichik and M. Yu. Antipin, J. Organomet. Chem., 1991, 406, 343–352 CrossRef.
- (a) M. F. Lappert, A. Singh, J. L. Atwood and W. E. Hunter, J. Chem. Soc., Chem. Commun., 1983, 206–207 RSC; (b) A. V. Khvostov, V. V. Nesterov, B. M. Bulychev, A. I. Sizov and M. Yu. Antipin, J. Organomet. Chem., 1999, 589, 222–225 CrossRef CAS; (c) D. Deng, X. Zheng, C. Qian, J. Sun and L. Chang, J. Organomet. Chem., 1994, 466, 95–100 CrossRef CAS.
- H. Schumann, M. R. Keitsch, J. Demtschuk and S. Műhle, Z. Anorg. Allg. Chem., 1998, 624, 1811–1818 CrossRef CAS.
- P. Zink, M. Visseaux and A. Mortreux, Z. Anorg. Allg. Chem., 2006, 632, 1943–1944 CrossRef CAS.
- S. M. Cendrowski-Guillaume, M. Nierlich, M. Lance and M. Ephritikhine, Organometallics, 1998, 17, 786–788 CrossRef CAS.
- (a) D. M. Barnhart, D. L. Clark, J. C. Gordoo, J. C. Huffman, R. L. Vincent, J. C. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 3487–3497 CrossRef CAS; (b) Y. Yao, X. Xu, B. Liu, Y. Zhang, Q. Shen and W.-T. Wong, Inorg. Chem., 2005, 44, 5133–5140 CrossRef CAS.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- F. M. Kerton, S. Holloway, A. Power, R. G. Soper, K. Sheridan, J. M. Lynam, A. C. Whitwood and C. E. Willans, Can. J. Chem., 2008, 86, 435–443 CrossRef CAS.
- (a) B. M. Chamberlain, Y. Sun, J. R. Hagadorn, E. W. Hemmesch, V. G. Young, Jr., M. Pink, M. A. Hillmyer and W. B. Tolman, Macromolecules, 1999, 32, 2400–2402 CrossRef CAS; (b) M. H. Chisholm and N. W. Eilerts, Chem. Commun., 1996, 853–854 RSC.
- G. M. Sheldrick, SHELXTL v. 6.12, Structure Determination Software Suite, Bruker AXS, Madison, Wisconsin, USA, 2000 Search PubMed.
- G. M. Sheldrick, SADABS v.2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
Footnote |
| † CCDC reference numbers 769020–769022. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0nj00486c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |