Inhibition of peroxidase-catalyzed iodination by thioamides: experimental and theoretical study of the antithyroid activity of thioamides†
Ghada J.
Corban‡
a,
Sotiris K.
Hadjikakou
*a,
Athanasios C.
Tsipis
a,
Maciej
Kubicki
b,
T.
Bakas
c and
Nick
Hadjiliadis
*a
aSection of Inorganic and Analytical Chemistry, Department of Chemistry, University of Ioannina, 45110 Ioannina, Greece. E-mail: shadjika@uoi.gr, nhadjis@uoi.gr; Fax: +30 26510-08786; Tel: +30 26510-08374, +30 26510-08420
bDepartment of Chemistry, A. Mickiewicz, University, ul. Grunwaldzka 6, 60-780 Poznan, Poland
cPhysics of Material Laboratory, Department of Physics, University of Ioannina, Greece
First published on 20th October 2010
Abstract
The reaction of di-iodine with N-methyl-2-mercapto-benzothiazole (NMBZT) in the presence of ferric tetra-phenyl-porphyrin chloride (FeTPPCl) in 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶3∶1 (I2∶thioamide∶FeTPPCl) molar ratio yields a mixture of products consisting of the iodonium {[(NMBZT)2I]+·[(I7)−]}n salt, 1, and the FeTPPCl complex, 2. The compounds were characterized by X-ray diffraction analysis. The crystal structure of 1 was identical to the one already published, while of compound 2 at 294(1) K re-evaluates previous work on it. The inhibition activity of the thioamide ligands (N-methyl-2-mercapto-benzothiazole (NMBZT), 2-mercapto-benzothiazole (MBZT), 5-chloro-2-mercapto benzothiazole (CMBZT), 2-mercaptothiazolidine (MTZT), 2-mercaptopyridine (PySH), thiourea (TU), 1,3-bis(3-pyridyl-methyl)-2-thiourea (PyTU), 2-mercapto-3,4,5,6-tetrahydropyrimidine (THP), 2-mercaptopyrimidine (PmSH), 6-propyl-2-thiouracil (PTU), 6-methyl-2-thiouracil (MTU), di-thiouracil-2,4 (DTUC), 2-mercapto-4-methyl-pyrimidine hydro chloride (MPmCl), methimazole (MMI), 2-mercapto-benzimidazole (BZIM), 5-nitro-2-mercapto-benzimidazole (NiMBZIM), 5-methyl-2-mercapto-benzimidazole (MBZIM), 2-hydroxy-pyrimidine (PmOH), 2-hydroxypyridine (PyOH)) against the catalytic oxidation of iodides by H2O2 in the presence of FeTPPCl (a model of the active site of Thyroid Peroxidase (TPO)) was measured as a result of the yield of I3− resulting from the oxidation of I− and was also evaluated theoretically using electronic structure calculation methods (DFT). The compounds exhibiting high and intermediate degree of inhibition (between 27% and 17%) are MBZT, MMI, PySH, NMBZT, PmSH, MBZIM, NiBZIM and MTZT. The kinetic study of inhibition of lactoperoxidase (LPO) (a model of TPO) is based on the assessment of LPO activity in the presence of its inhibitors—the thioamide compounds. LPO activity can be assayed by measuring guaiacol (gua) peroxidation to tetraguaiacol in the presence of H2O2. The results illustrated that the lowest IC50 values (<10 μM) required for the LPO inhibition were exhibited by DTUC, NiMBZIM, MBZT, PySH thioamides. Low concentrations between 10 and 30 μM are obtained in the case of CMBZT, BZIM, MPmCl, PmSH, MBZIM, and MMI. DFT calculations of the geometric and energetic profiles threw light on the mechanism of the inhibition process by the thioamide compounds scrutinizing all crucial reaction steps and are in agreement with the experimental results.
∶3∶1 (I2∶thioamide∶FeTPPCl) molar ratio yields a mixture of products consisting of the iodonium {[(NMBZT)2I]+·[(I7)−]}n salt, 1, and the FeTPPCl complex, 2. The compounds were characterized by X-ray diffraction analysis. The crystal structure of 1 was identical to the one already published, while of compound 2 at 294(1) K re-evaluates previous work on it. The inhibition activity of the thioamide ligands (N-methyl-2-mercapto-benzothiazole (NMBZT), 2-mercapto-benzothiazole (MBZT), 5-chloro-2-mercapto benzothiazole (CMBZT), 2-mercaptothiazolidine (MTZT), 2-mercaptopyridine (PySH), thiourea (TU), 1,3-bis(3-pyridyl-methyl)-2-thiourea (PyTU), 2-mercapto-3,4,5,6-tetrahydropyrimidine (THP), 2-mercaptopyrimidine (PmSH), 6-propyl-2-thiouracil (PTU), 6-methyl-2-thiouracil (MTU), di-thiouracil-2,4 (DTUC), 2-mercapto-4-methyl-pyrimidine hydro chloride (MPmCl), methimazole (MMI), 2-mercapto-benzimidazole (BZIM), 5-nitro-2-mercapto-benzimidazole (NiMBZIM), 5-methyl-2-mercapto-benzimidazole (MBZIM), 2-hydroxy-pyrimidine (PmOH), 2-hydroxypyridine (PyOH)) against the catalytic oxidation of iodides by H2O2 in the presence of FeTPPCl (a model of the active site of Thyroid Peroxidase (TPO)) was measured as a result of the yield of I3− resulting from the oxidation of I− and was also evaluated theoretically using electronic structure calculation methods (DFT). The compounds exhibiting high and intermediate degree of inhibition (between 27% and 17%) are MBZT, MMI, PySH, NMBZT, PmSH, MBZIM, NiBZIM and MTZT. The kinetic study of inhibition of lactoperoxidase (LPO) (a model of TPO) is based on the assessment of LPO activity in the presence of its inhibitors—the thioamide compounds. LPO activity can be assayed by measuring guaiacol (gua) peroxidation to tetraguaiacol in the presence of H2O2. The results illustrated that the lowest IC50 values (<10 μM) required for the LPO inhibition were exhibited by DTUC, NiMBZIM, MBZT, PySH thioamides. Low concentrations between 10 and 30 μM are obtained in the case of CMBZT, BZIM, MPmCl, PmSH, MBZIM, and MMI. DFT calculations of the geometric and energetic profiles threw light on the mechanism of the inhibition process by the thioamide compounds scrutinizing all crucial reaction steps and are in agreement with the experimental results.
Introduction
The interest in the study of structure–activity relationship of thioamides against di-iodine is stimulated by the interest in the molecular compounds formed between antithyroid drugs and di-iodine,1–13 since thioamides are known to exhibit anti-thyroid activity.13 The most commonly employed anti-thyroid drugs in use are 6-n-propyl-thiouracil (PTU), N-methyl-imidazoline-2-thione (methimazole, MMI) and 3-methyl-2-thioxo-4-imidazoline-1-carboxylate (carbimazole) (CBZ).13 Thyroid-peroxidase (TPO), an iron–porphyrin enzyme14,15 is responsible for the oxidation of iodide anions to active di-iodine.15–19 The proposed reaction scheme for the mechanism of action of TPO involves the formation of the {[TPO–OI]−} intermediate complex which reacts with tyrosine to give mono- and di-iodo-tyrosine and TPO15,19 (Scheme 1). | ||
| Scheme 1 The TPO catalyzed iodination of tyrosyl residues of thyroglobuline. | ||
Thioamides exhibiting anti-thyroidal activity against the hyper-thyroidism (Graves' disease) can be classified into two categories: (i) thioamides which react with di-iodine, forming mono-, di-cationic salts, like MMI1–9,20 and (ii) thioamides able to form weak charge transfer compounds with di-iodine, like PTU.2 Thus, it seems that while drugs like MMI etc., proposed to interfere with the iodination mechanism in their ability to form the active iodine species, compete with tyrosyl residues of thyroglobuline for the active iodine;1–9,20–23 drugs like PTU may function instead by their ability to inhibit the activity of the thyroid-peroxidase (TPO) or the iodothyronine deiodinase (ID-1), an enzyme responsible for the monodeiodination of the T4 prohormone to the T3 hormone. N-Methyl-2-mercapto-benzothiazole (NMBZT) may also be active similar to PTU since it forms weak CT complexes with di-iodine with “spoke” structures.5
We had previously shown that when NMBZT reacts with di-iodine in the presence of FeCl3 in 6∶3∶1 ratio (I2∶NMBZT∶FeCl3) the complexes {[(NMBZT)2I]+}·[FeCl4]− and {[(NMBZT)2I]+}·[I7]− were formed.5 In the course of our studies on the interaction of thioamides with di-iodine (I2) we have investigated the reaction of di-iodine with N-methyl-2-mercapto-benzothiazole (NMBZT) in the presence of ferric tetra-phenyl-porphyrin chloride (FeTPPCl). This reaction results in the formation of a mixture of products consisting of (i) the iodonium salt of formula {[(NMBZT)2I]+·[(I7)−]}n, 1, and (ii) the starting compound FeTPPCl 2. The products were characterized by X-ray diffraction analysis. In addition the inhibition activity of the thioamide ligands (Chart 1) towards the catalytic oxidation of iodides by H2O2 in the presence of FeTPPCl (a model of the active site of Thyroid Peroxidase (TPO)) was also measured experimentally and evaluated theoretically based on DFT calculations. Furthermore a kinetic study of the inhibition of lactoperoxidase (LPO) (a model of TPO) was based on the determination of LPO activity in the presence of its inhibitors—the thioamide compounds.19
 | ||
| Chart 1 | ||
Results and discussion
Reactions
Since TPO contains iron porphyrin in its active site we decided to extend our studies and investigate the reactivity of di-iodine of the NMBZT in the presence of FeTPPCl which is the active site of TPO. The results obtained are described by reaction (1), where NMBZT can be any thioamide used in this study (see details below)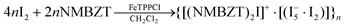 | (1) |
An ORTEP diagram of the crystal structure of compound 2 is shown in Fig. 1, while selected bond distances and angles are collected in Table 1
 | ||
| Fig. 1 (A) Molecular diagram together with labeling scheme of FeTPPCl (2). Thermal ellipsoids drawn at the 50% probability level. FeTPPCl is a mixture of two superimposed different structures (A) and (B). | ||
| Molecule A | Molecule B | ||
|---|---|---|---|
| 1 Symmetry transformations used to generate equivalent atoms: a = −x, −1/2 + z. b = 2 − x, −y, z. c = 1 + y, 1 − x, z. d = 1/2 + x, −1/2 + y, 1/2 + z. k = 1/2 + y, 1/2 − x, −1/2 + z. l = −1/2 + x, 1/2 + y, 1/2 + z. m = 1/2 − y, −1/2 + x, 1/2 + z. n = 1/2 + y, 3/2 − x, 1/2 + z. | |||
| Bond lengths (Å) | |||
| Fe(1A)–N(1) | 2.0732(19) | Fe(1B)–N(1) | 2.0399(18) |
| Fe(1A)–Cl(1A) | 2.140(3) | Fe(1B)–Cl(1B) | 2.288(6) |
| N(1)–C(2) | 1.380(3) | N(1)–C(4)#2 | 1.381(3) |
| C(2)–C(3) | 1.399(3) | C(2)–C(6)#2 | 1.433(3) |
| C(3)–C(4) | 1.403(3) | C(3)–C(31) | 1.491(3) |
| C(31)–C(32) | 1.349(6) | C(31)–C(36) | 1.387(6) |
| C(32)–C(33) | 1.429(7) | C(33)–C(34) | 1.368(8) |
| C(34)–C(35) | 1.362(7) | C(35)–C(36) | 1.347(6) |
| C(4)–N(1)#3 | 1.381(3) | C(4)–C(5) | 1.440(3) |
| C(5)–C(6) | 1.359(3) | C(6)–C(2)#3 | 1.433(3) |
| Bond angles (°) | |||
| N(1)#1–Fe(1A)–N(1) | 154.1(2) | N(1)–Fe(1B)–N(1)#1 | 164.2(3) |
| N(1)#2–Fe(1A)–N(1) | 87.12(5) | N(1)–Fe(1B)–N(1)#2 | 88.92(4) |
| N(1)–Fe(1A)–Cl(1A) | 102.95(11) | N(1)–Fe(1B)–Cl(1B) | 97.91(14) |
| C(2)–N(1)–C(4)#2 | 105.95(15) | C(2)–N(1)–Fe(1B) | 126.83(13) |
| C(4)#2–N(1)–Fe(1B) | 126.58(13) | C(2)–N(1)–Fe(1A) | 125.81(14) |
| C(4)#2–N(1)–Fe(1A) | 125.76(15) | N(1)–C(2)–C(3) | 125.93(18) |
| N(1)–C(2)–C(6)#2 | 109.98(17) | C(3)–C(2)–C(6)#2 | 124.06(19) |
| N(1)#3–C(4)–C(5) | 109.79(18) | N(1)#3–C(4)–C(3) | 126.01(17) |
| Torsion angles (°) | |||
| N(1_a)–Fe(1A)–Fe(1B)–N(1) | 90.00(7) | N(1_b)–Fe(1A)–Fe(1B)–N(1) | 180.0(3) |
| Fe(1B)–Fe(1A)–N(1)–C(2) | 100.5(4) | N(1)–Fe(1B)–N(1_c)–Fe(1A) | 2.15(14) |
| Cl(1A)–Fe(1A)–N(1)–Fe(1B) | 180.0(3) | Cl(1B)–Fe(1B)–N(1)–Fe(1A) | −180.00(1) |
| N(1_b)–Fe(1A)–N(1)–Fe(1B) | 0.00(15) | N(1_c)–Fe(1A)–N(1)–Fe(1B) | 77.38(11) |
| Cl(1A)–Fe(1A)–N(1_a)–C(4) | 80.1(4) | Cl(1B)–Fe(1B)–N(1_a)–C(4) | −84.4(4) |
| N(1)–Fe(1A)–N(1_a)–C(4) | −22.5(4) | N(1_a)–Fe(1B)–N(1)–C(2) | −12.9(4) |
The FeTPPCl compound, 2, had already been published by Fleischer et al.25 followed by Hoard et al. (B),26 who analyzed it as aquahydroxyiron(III) tetraphenylporphyrin [(H2O)Fe(OH)(TPP)]. Hoard et al.26 reported the structure as disordered across the mirror plane in the space group I4/m (a = 13.53, c = 9.82 Å), consisting of two anti-parallel superimposed molecules: the TPP fragment was on the mirror plane and the Fe and Cl atoms are symmetrically disposed on either side of this plane. The symmetry requires the equal occupation of both alternatives. The space group was however left open to more consideration between the following choices I4, I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) , and I4/m.26
, and I4/m.26
Our data for compound 2 were collected at 293 K and they confirm the tetragonal symmetry with unit cell dimensions a = 13.5590(19), c = 9.836(2) Å. We have chosen however the description in the space group I4 with two alternative positions of Cl and Fe atoms freely refined above and below the TPP plane. We have also tried the I4/m refinement and the R factors were by ca. 1.5% higher without the significant advantage in e.g. thermal ellipsoids, as well as of I, with worst results.
Consequently, the two superimposed structures have different distances between the iron atom and the plane defined by the four N atoms. In molecule A the iron atom is displaced by 0.476 Å from the plane defined by the four N atoms, while in B iron lies out-of-plane by 0.284 Å, whereas in the case of the reported structure this distance is 0.383(5) Å.26 The Fe(1)–Cl(1) is 2.148(4) in (A) and 2.260(6) in (B) compared to the 2.192 (12) Å for the structure reported.26 As for the Fe–N bond distances are equal (by symmetry) in the same molecule but vary between 2.0660(17) for (A) and 2.0429(15) for (B) compared to 2.049(9) Å in the reported structure.26
The Mössbauer spectrum of FeTPPCl (Fig. 2) gives one signal for Fe(III) in liquid helium temperature of 5 K. However, in liquid nitrogen at a temperature of 80 K there is a broadening of the signal attributed to the two different superimposed iron centers of FeTPPCl.
 | ||
| Fig. 2 Mossbauer spectra of 2 at 5 and 80 K. | ||
We further investigated Scheme 1,using reaction (1), by varying the thioamide ligands, as follows:
Inhibition of the catalytic activity of FeTPPCl
Iodide anion (I−) is easily oxidized by H2O2 in a 2e− transfer reaction either non-enzymatically or enzymatically in the presence of peroxidase, according to the following equation:27| 2 I− + H2O2 → I2 + H2O | (2) |
| I− + I2 ⇌ I3− | (3) |
Oxidation of iodide anion by hydrogen peroxide
The inhibition activity of the thioamides and other ligands was measured in the presence of FeTPPCl (a model of the active site of TPO14,15) and H2O2 yielding I3− resulting from the oxidation of I−27 according to reaction (3). The ligands involved in this study were: N-methyl-2-mercapto-benzothiazole (NMBZT), 2-mercapto-benzothiazole (MBZT), 5-chloro-2-mercapto benzothiazole (CMBZT), 2-mercaptothiazolidine (MTZT), 2-mercaptopyridine (PySH), thiourea (TU), 1,3-bis(3-pyridyl-methyl)-2-thiourea (PyTU), 2-mercapto-3,4,5,6-tetrahydropyrimidine (THP), 2-mercaptopyrimidine (PmSH), 6-propyl-2-thiouracil (PTU), 6-methyl-2-thiouracil (MTU), di-thiouracil-2,4 (DTUC), 2-mercapto-4-methyl-pyrimidine hydro chloride (MPmCl), methimazole (MMI), 2-mercapto-benzimidazole (BZIM), 5-nitro-2-mercapto-benzimidazole (NiMBZIM), 5-methyl-2-mercapto-benzimidazole (MBZIM), 2-hydroxy-pyrimidine (PmOH), and 2-hydroxypyridine (PyOH).
The catalytic mechanism of FeTPPCl could be explained as follows: first FeTPPCl is oxidized by H2O2 liberating H2O (Scheme 1). The oxidized form then reacts with I− to yield the [FeTPPOI]− intermediate which in the presence of excess I− regenerates the catalyst (FeTPPCl) affording first I2 which is converted to I3−. If [FeTPPOI]− reacts with H2O then it regenerates the catalyst and gives I− and consequently I3− (Scheme 1). Di-iodine could not be detected spectroscopically in this study because once formed, its transformation to the tri-iodide anion, in the presence of excess I− is instantaneous, in combination with its absorption in aqueous solutions which is negligible at the wavelengths maxima of I3−.28
The inhibition of the catalytic activity of FeTPPCl could probably be due to the interaction of the thioamide ligands with the [FeTPPOI]− intermediate. This leads to the formation of dead end products; moreover it renders the intermediate unavailable to further react in order to regenerate the FeTPPCl catalyst. If the thioamides react with the oxidized form of FeTPPCl, various oxidation products could be obtained. However, this step is not important since in the presence of excess I−, the [FeTPPOI]− intermediate is dominantly formed.
The absorption spectra of I3− species show maxima at 288 nm and 353 nm.28 The formation of the I3− species was assayed by its absorbance at 353 nm and its molar absorption coefficient ε = 25550 M−1 cm−1.28,29 Measurements were performed at room temperature using solutions in a mixture of dichloromethane and methanol 60∶40% by volume. It should be noticed that the use of the mixture of CH2Cl2 and MeOH solvents help in protecting the iron porphyrin30 and in maintaining the tri-iodide mixture equilibrium (3) thus preventing the extinction coefficient from varying.27
The assay system contained KI = 10 mM (160 μl of 0.125 M KI), [H2O2] = 50 μM (10 μl of 30% w/v HO), [FeTPPCl] = 5 μM (10 μl of 10−3 M FeTPPCl), [L] = 5 μM (10 μl of 10−3 M thioamide). The volume was adjusted to the total of 2 ml in the cell. The use of an excess of iodide is to ensure the complete conversion of all the di-iodine formed from oxidation of iodide by H2O2 into tri-iodide.17 Also I− is needed in excess since below a concentration of 10 mM no I3− or I2 production could be detected spectroscopically.31
The reaction of KI in the presence of H2O2 gave 50% I3−, while in the presence of the catalyst the yield was 98%. The amount of I3− present was calculated from the spectra using the fact that the molar ratio between H2O2 degradation and I3− formation was 1∶1 (eqn (2)).17,27 Hence, a 100% production of I3− is equivalent to a concentration of 50 μM of I3− which is the maximum amount of H2O2 consumed in this reaction. The percent inhibition was calculated from the difference between the reference (98%) and the percent of I3− formed in the presence of the thioamide ligands.
In order to make sure that the ligands do not absorb at 353 nm, the spectra of the ligands with FeTPPCl only were recorded and compared with the spectrum of KI + H2O2 in the absence of FeTPPCl. In this way it was proven that the catalytic activity was solely due to FeTPPCl and that the thioamide ligands decreased the activity of the porphyrin catalyst, while they did not affect the oxidation of I−. Thioamide ligands showing absorptions overlapping with the identification peak of I3− at 353 nm were excluded from this study. The spectrum of FeTPPCl were recorded with and without H2O2 and the absorption at 353 nm was found to be negligible as compared to that of I3−, formed in the presence and absence of FeTPPCl (Fig. 3A).
 | ||
| Fig. 3 (A) UV spectra of FeTPPCl with and without H2O2 compared to I3− formation. (B) UV spectra of inhibition of FeTPPCl by MBZT, NMBZT, PySH and MMI, compared to the controls at 353 nm. | ||
Fig. 3B shows the spectra of MBZT, NMBZT, PySH and MMI ligands and their effect in reducing the catalytic activity of FeTPPCl when added to the assay system containing iodide, hydrogen peroxide and the porphyrin catalyst.
Table 2 shows the percent of inhibition caused by the compounds under study with the confidence limit. Three categories can be identified according to the relative values of the degree of inhibition caused by the different ligands: strong, intermediate and very low. The compounds that show a high degree of inhibition (between 27% and 19%) are MBZT, MMI, PySH, PmSH and MBZIM. These compounds form either ionic or neutral disulfide complexes with I21–9 except for MBZT which forms charge transfer complexes with di-iodine.
| Thioamide | % Inhibition | Confidence |
|---|---|---|
| 1 NMBZT = N-methyl-2-mercapto-benzothiazole, MBZT = 2-mercapto-benzothiazole, PySH = 2-mercaptopyridine, PmOH = 2-hydroxy-pyrimidine, MMI = methimazole, NiMBZIM = 5-nitro-2-mercapto-benzimidazole, TU = thiourea, PmSH = 2-mercaptopyrimidine, MBZIM = 2-mercapto-benzimidazole, MBZIM = 5-methyl-2-mercapto-benzimidazole, MTZT = 2-mercaptothiazolidine, THP = tetrahydropyrimidine, PyOH = 2-hydroxypyridine, PTU = propyl thiouracil, PyTU = 1,3-bis(3-pyridyl-methyl)-2 thiourea, CMBZT = 5-chloro-2-mercapto benzothiazole, MPmCl = 2-mercapto-4 methyl-pyrimidine hydro chloride | ||
| MBZT | 27.11 | 0.62 |
| MMI | 25.48 | 0.86 |
| PySH | 21.87 | 1.48 |
| NMBZT | 20.20 | 0.70 |
| PmSH | 19.54 | 1.33 |
| MBZIM | 19.05 | 0.72 |
| NiBZIM | 17.86 | 0.81 |
| MTZT | 16.80 | 0.61 |
| PmOH | 15.78 | 0.84 |
| TU | 10.98 | 1.14 |
| MeBZIM | 10.24 | 0.86 |
| PyOH | 9.51 | 0.55 |
| PTU | 7.81 | 0.77 |
| PyTU | 7.09 | 0.38 |
| THP | 5.61 | 0.55 |
| CMBZT | 3.95 | 1.15 |
| MPmCl | 1.31 | 0.78 |
The other set of compounds with intermediate inhibition % includes the ligands of NiBZIM, MTZT, PmOH, TU and MeBZIM. These ligands form ionic salt complexes with di-iodine. Exception is NiBZIM, which undergoes de-sulfuration in the presence of I2.
NMBZT shows also a relatively high inhibition (20.2%). The crystal structure of NMBZT occurs in two forms the spoke structure [LI2] and the iodonium salt 1.5 Moreover, it forms also an ionic compound formulated as {[L2I]+[FeCl4−]},5 in the presence of FeCl3. Therefore, its high degree of inhibition can be attributed to its ionic interaction through formation of [L2I]+ with the iron center of the catalyst FeTPPCl.
2-Hydroxy-pyrimidine (PmOH) does not contain a sulfur donor atom to be able to form charge transfer complexes with di-iodine, but exhibits an intermediate degree of inhibition. This could be attributed to the ionic nature of the ligand [(PmOH2)]+Cl− in addition to the ionic compound it forms when interacting with di-iodine {[(C4H5N2O)+]2·[I2Cl2]2−}.9 The compounds that show very low inhibition are PyOH, PTU, PyTU, THP, CMBZT and MPmCl. The crystal structures of PTU and CMBZT showed that these compounds can only form spoke structures with I2 of formula [LI2]2 with different stoichiometry in the presence or absence of an iron center. No crystal structures are known for MPmCl and for PyTU. These thioamides showed either a very low inhibition or almost no inhibition at all.
Computational insights on the mechanism of inhibition of the catalytic activity of FeTPPCl
Consistent with the X-ray crystal structures of the compounds formed by the interaction of the antithyroid drugs MMI and PTU (Fig. 4) with I2, the computed sulfur–diiodine bond dissociation energies, BDE(S–I2), in the charge transfer complexes (CT) of MMI–I2 and PTU–I2 were found to be only 12.2 and 7.0 kcal mol−1, respectively, at the BP86/6-31G(d,p) ∪ Def2-QZVPP(I) level. On the other hand, in the cationic MMI–I+–MMI or PTU–I+–PTU iodonium species, the computed sulfur–I+ bond dissociation energy, BDE(S–I+), was found to be much higher which amounted to 30.8 and 25.0 kcal mol−1 respectively. The low BDE(S–I2) of PTU–I2 compared to the ionic compound indicates that the formation of the CT spoke structure is energetically favored; in fact PTU despite our efforts was only found to form the spoke [PTU–I2] structure2 when combined to di-iodine in the presence or absence of iron containing compounds. The spoke structure of MMI is also energetically feasible to exist; however in the solid state MMI exists in the monocationic disulfide [{(C4H6N2S–SN2H5C4)2+}·(I3)−·(I5)−] (C4H6N2S = MMI) and dicationic disulfide [{(C4H5N2S–SN2H5C4)2+}·(I8)2−].20 The existence of MMI as a disulfide with iodide counter anions does not conflict the estimated BDE values, since it is well-established that the disulfide intermediates formed have the spoke structure7 and the MMI–I2 complex exists in solution.20 On the other hand, the relatively high-energy values of MMI–I+–MMI or PTU–I+–PTU species suggest that they are not likely to exist. In effect, the MMI–I+–MMI or PTU–I+–PTU species have not been isolated up to now.![Equilibrium geometries of MMI, MMI·I2, [MMI–I–MMI]+, PTU, PTU·I2 and [PTU–I–PTU]+ along with selected bond distances (Å) and angles (°) computed at the BP86/6-31G(d,p)(E) ∪ Def2-QZVPP(i) level of theory.](/image/article/2011/NJ/c0nj00626b/c0nj00626b-f4.gif) | ||
| Fig. 4 Equilibrium geometries of MMI, MMI·I2, [MMI–I–MMI]+, PTU, PTU·I2 and [PTU–I–PTU]+ along with selected bond distances (Å) and angles (°) computed at the BP86/6-31G(d,p)(E) ∪ Def2-QZVPP(I) level of theory. | ||
The [Fe(TPP){O–I⋯S(thione)}] acceptor–donor interactions predicted to be frontier-orbital controlled involve the interaction of the donor frontier molecular orbitals (FMOs) of MMI or PTU with the acceptor FMO of the [FeTPP(OI)] (4A) complex (Fig. S1, ESI†).
Noteworthy is the formation of a relatively weak O–I bond in [Fe(TPP)(OI)] (4A), the predicted bond dissociation energy for the homolytic dissociation of the O–I bond being 18.1 kcal mol−1 and therefore the Fe–O–I⋯S(thione) interaction is thermodynamically favored. The estimated Wiberg Bond Index (WBI) of 1.007 indicates that the O–I bond corresponds to a single bond. The iodide ligand in [Fe(TPP)(OI)] (4A) acquires a positive natural atomic charge of 0.30 |e|, while the oxygen atom acquires a negative natural atomic charge of −0.65 |e|. Moreover, the total spin density is distributed on the Fe–O–I framework, the Mulliken spin atomic densities being 2.34, 0.39 and 0.25 for the Fe, O and I atoms, respectively.
The mechanism of the inhibition of the catalytic activity of FeTPPCl by the thioamide compounds has been investigated for both the quartet and sextet potential energy surfaces considering the following reaction path:
Initially the FeTPPCl complex interacts with H2O2 to form the FeCl(TPP)(κ1O-H2O2) intermediate followed by elimination of H2O to yield the FeTPPCl(O) intermediate which upon reaction with iodide anion I− affords the [Fe(TPP)(OI)]− complex. This is crucial for the iodination of tyrosyl residues of thyroglobuline. The optimized structures of all stationary points located on the quartet and sextet potential energy surfaces of the inhibition of the catalytic activity of FeTPPCl by the thioamide compounds following the aforementioned reaction pathway are displayed in Fig. S2 (ESI†), while the spin density distributions are depicted schematically in Fig. S3 (ESI†).
Relative energies calculated with respect to the energy of FeTPPCl (4A1) plus the energy of H2O2 for the quartet PES and the energy of FeTPPCl (6A2) plus the energy of H2O2 for the sextet PES are given in the energy and geometric profiles shown in Fig. 5.
 | ||
| Fig. 5 Energetic profile of the inhibition of the catalytic activity of FeTPPCl by the thioamide compounds for both the quartet and sextet potential surfaces (PESs) computed at the UBP86/Def2-QZVPP(Fe,I) ∪ 6-31G(d,p)(E) level of theory. | ||
The intermediate-spin (4A1) structure of FeTPPCl was found to be the lowest energy complex. The high-spin (6A2) structure of FeTPPCl corresponds to a local minimum of 8.3 kcal mol−1 higher in energy than the 4A1 ground state. The low-spin (2A1) structure was not possible to be located on the PES, due to SCF convergence problems. Note that a doublet spin state for a five-coordinate Fe3+ complex appears to be rather artificial. In the FeTPPCl (4A1) structure with C4v symmetry the central metal ion resides 0.296 Å above the plane of the four nitrogen atoms and the macrocyclic ligand is perfectly planar. This value is very close to the experimentally estimated value of 0.284 Å for structure B of FeTPPCl (Fig. 1). In general the UBP86/Def2-QZVPP(Fe,I) ∪ 6-31G(d,p)(E) optimized geometry of FeTPPCl (4A1) in the gas phase closely resembles structure B determined by X-ray crystallography (Fig. 1). On the other hand, the UBP86/Def2-QZVPP(Fe,I) ∪ 6-31G(d,p)(E) optimized geometry of FeTPPCl (6A2) in the gas phase closely resembles structure A determined by X-ray crystallography (Fig. 1). In the FeTPPCl (6A2) structure with C4v symmetry the central metal ion resides 0.510 Å above the plane of the four nitrogen atoms, a value very close to the experimental value of 0.476 Å. It is clear that the superimposed structures result from crystallization of the intermediate-spin 4A1 and high-spin 6A2 structures coexisting in equilibrium in solution. The spin density in complexes FeTPPCl (4A1) and FeTPPCl (6A2) (Fig. S3, ESI†) is mainly located on iron (2.50 and 3.98 respectively) and to a lesser extent to the chloride ligand (0.40 and 0.38 respectively). In the high-spin FeTPPCl (6A2) complex a sizeable spin delocalization on the macrocyclic ligand is indicated by a total spin density of 0.44, mainly localized on the four nitrogen donor atoms.
The FeTPPCl (4A1) and FeTPPCl (6A2) complexes upon reacting with hydrogen peroxide yield hydrogen peroxide adducts FeTPPCl(κ1-O(H)OH) (4A) and FeTPPCl(κ1-O(H)OH) (6A), respectively, where the H2O2 ligand coordinates “end-on” to the iron center in a κ1-O(H)OH bonding mode.15,19 The intermediate-spin FeTPPCl(κ1-O(H)OH) (4A) complex was found to be the ground state structure, while the high-spin FeTPPCl(κ1-O(H)OH) (6A) complex is a local minimum of 8.6 kcal mol−1 higher in energy than the ground state. The Fe–O2H2 distances are 2.683 and 2.769 Å in FeTPPCl(κ1-O(H)OH) (4A) and FeTPPCl(κ1-O(H)OH) (6A), respectively, while the central iron atom resides 0.247 and 0.432 Å out of the plane of the four nitrogen atoms of the macrocyclic ligand towards the axial chloride ligand respectively. In both adducts, the Fe–O coordination bond is very weak, with an estimated interaction energy around 3–4 kcal mol−1. In practice such interaction energies are comparable to weak hydrogen bond energies. In effect in both adducts hydrogen bonds are formed between both H atoms of the coordinated H2O2 ligand and the negatively charged adjacent nitrogen atoms of the macrocyclic ligand (Fig. S2, ESI†), the H⋯N distances found in the range of 1.993–2.885 Å. The estimated WBI(Fe–O) value of 0.18 indicates marginal Fe⋯O interactions as well. The spin density distribution in the FeTPPCl(κ1-O(H)OH) (4A) and FeTPPCl(κ1-O(H)OH) (6A) complexes is similar to that of their precursor FeTPPCl (4A1) and FeTPPCl (6A2) complexes respectively (Fig. S3, ESI†).
In the next step, elimination of water from the coordinated H2O2 ligand yields the [FeTPPCl(O)]− (3A1) and [FeTPPCl(O)]− (5A2) complexes. The water elimination step is predicted to be exothermic, the estimated exothermicities are found to be −91.2 and −80.7 kcal mol−1 on the quartet and sextet PESs, respectively, at the UBP86/Def2-QZVPP(Fe,I) ∪ 6-31G(d,p)(E) level. The next step involving the reaction of the [FeTPPCl(O)]− (3A1) and [FeTPPCl(O)]− (5A2) complexes with the iodide anion to form the [FeTPPCl(OI)]− (4A) and [FeTPPCl(OI)]− (6A) complexes, respectively, is endothermic, with the endothermicity predicted to be 54.9 and 51.3 kcal mol−1 on the quartet and sextet PESs respectively. In the following step, elimination of the chloride ligand from the [FeTPPCl(OI)]− (4A) and [FeTPPCl(OI)]− (6A) complexes yields the FeTPP(OI) complexes with an energy demand of 28.7 and 23.0 kcal mol−1 for the quartet and sextet PESs respectively.
The low-spin (3A1) structure of [FeTPPCl(O)]− was found to be the global minimum, with the high-spin (5A2) structure corresponding to a local minimum of 18.8 kcal mol−1 higher in energy than the 3A1 ground state. The equilibrium geometries of the [FeTPPCl(O)]− (3A1) and [FeTPPCl(O)]− (5A2) intermediates are octahedral with C4v symmetry. The computed structure of the Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety (Fe–O bond length of 1.665 and 1.661 Å) is comparable to that published recently by Decker and Solomon32 who showed that the geometry of the FeIVO subunit does not depend on the nature of the ligand, whether heme or non-heme in character. The estimated WBI(Fe–O) value of 1.55 indicates that the Fe–O has a partially double bond character. In the high-spin 3A2 state the iron central atom resides 0.085 Å out of the plane of the four nitrogen atoms of the macrocyclic ligand towards the axial oxo ligand. The spin density in complexes [FeTPPCl(O)]− (3A1) and [FeTPPCl(O)]− (5A2) (Fig. S3, ESI†) is mainly located on iron (1.22 and 3.05 respectively) and to a lesser extent to the oxo-ligand (0.83 and 0.70 respectively). In the high-spin [FeTPPCl(O)]− (5A2) complex a sizeable spin delocalization on the macrocyclic ligand is indicated by a total spin density of 0.24, mainly localized on the four nitrogen donor atoms.
O moiety (Fe–O bond length of 1.665 and 1.661 Å) is comparable to that published recently by Decker and Solomon32 who showed that the geometry of the FeIVO subunit does not depend on the nature of the ligand, whether heme or non-heme in character. The estimated WBI(Fe–O) value of 1.55 indicates that the Fe–O has a partially double bond character. In the high-spin 3A2 state the iron central atom resides 0.085 Å out of the plane of the four nitrogen atoms of the macrocyclic ligand towards the axial oxo ligand. The spin density in complexes [FeTPPCl(O)]− (3A1) and [FeTPPCl(O)]− (5A2) (Fig. S3, ESI†) is mainly located on iron (1.22 and 3.05 respectively) and to a lesser extent to the oxo-ligand (0.83 and 0.70 respectively). In the high-spin [FeTPPCl(O)]− (5A2) complex a sizeable spin delocalization on the macrocyclic ligand is indicated by a total spin density of 0.24, mainly localized on the four nitrogen donor atoms.
The intermediate-spin (4A) structure of [FeTPPCl(OI)]− corresponds to the global minimum, while the high-spin (6A) structure is a local minimum of 15.2 kcal mol−1 higher in energy than the quartet ground state. The optimized structures of the [FeTPPCl(OI)]− (4A) and [FeTPPCl(OI)]− (6A) intermediates correspond to octahedral geometries. The estimated WBI(O–I) value of 0.40 is indicative of weak O⋯I interactions corresponding to one electron bond. The spin density in the [FeTPPCl(OI)]− (4A) complex (Fig. S3, ESI†) is almost equally distributed on the O–I moiety (1.48) and the central iron atom (1.44). A smaller amount of spin density (0.24) is also located on the chloride ligand. In the high-spin [FeTPPCl(OI)]− (6A) complex the spin density is mainly located on iron (3.84) and to a lesser extent on the O–I moiety (0.59).
Finally, the intermediate-spin (4A) structure of FeTPP(OI) corresponds to the global minimum, with the high-spin (6A) structure being a local minimum 9.3 kcal mol−1 higher in energy than the quartet ground state. The optimized structures of the FeTPP(OI) (4A) and FeTPP(OI) (6A) complexes exhibit square pyramidal geometries with the O–I moiety at the apex of the pyramid. The estimated WBI(O–I) value of 1.00 indicates the formation of a single O–I bond. This is consistent with the shorter O–I bond length in the FeTPP(OI) complexes than the [FeTPPCl(OI)]− ones (1.987 vs. 2.409 and 1.972 vs. 1.999 Å for the quartet and sextet states respectively). The spin density in the quartet and sextet FeTPP(OI) complexes (Fig. S3, ESI†) is mainly located on the iron central atom (2.34 and 3.92 respectively) and to a lesser extent on the O–I moiety (0.64 and 0.56 respectively).
In summary, as is evident from the energy data the quartet and sextet pathways are almost equivalent with the former being slightly favored. The crucial intermediate intervening in the inhibition of the catalytic activity of FeTPPCl by thioamides corresponds to [FeCl(TPP)(OI)]− (Scheme 1). This intermediate is even more stable than the FeTPPCl complex. Moreover in agreement with the experimental results on FeTPPCl, PTU does not exhibit very low activity with respect to inhibition towards FeTPPCl, mainly because it's not able to form ionic or disulfide compounds. On the other hand, MMI forming ionic disulfides with di-iodine exhibits a high inhibition activity with respect to FeTPPCl. This observation vindicates the suggestion that the inhibition mechanism of the active site of TPO involves ionic interaction with the [E–OI]− intermediate.
Kinetic study of LPO
The kinetic study of inhibition of lactoperoxidase (LPO) is based on the assessment of LPO activity in the presence of its inhibitors—the thioamide compounds. LPO activity can be assessed by measuring guaiacol (gua) peroxidation to tetraguaiacol in the presence of H2O2 according to the following reaction:
Measurements of changes in the absorbance of tetraguaicol were recorded at 470 nm (ε = 2.66 × 104 M−1 cm−1)33,34
Before recording the kinetic spectra essential for collecting the data for this study, it was necessary to run the spectrum of the enzyme (LPO) and the spectra of the enzyme with the substrate (gua) in the absence and presence of H2O2. These measurements aimed in observing the different products as well ensuring that no absorption other than the oxidation of guaiacol takes place at 470 nm (Fig. 6A).
 | ||
| Fig. 6 (A) Spectra of LPO, with the substrates with and without H2O2. (B) Variation of the spectra of LPO–H2O2 system at 40 min interval. Each spectrum has been recorded after 5 min from the previous. (C) Variation of the spectra of LPO–H2O2–guaiacol system at 20 min interval. Each spectrum has been recorded after 5 min later to the previous one. | ||
The spectrum of LPO in the presence of H2O2 was taken at an interval time of 40 min with the first spectrum being at time zero and then consecutively by recording it every 5 min (Fig. 6B). The native LPO enzyme has its absorption maxima at 412, 500, 542, 590 and 630 nm.35 The spectra of the LPO–H2O2 system follow at a λ < 280 nm the order (Fig. 6B, starting from below): native LPO, initial (at time zero [t0] of adding H2O2), 5 min, increasing until we reach the final time (40 min). Between 280 and 305 nm many individual isosbestic points exist between two consecutive spectra, however the sharp isosbestic point between all the spectra exist at 306 nm except for the native enzyme and the initial measurement. The initial spectra have their first isosbestic point at 316 nm, and that of the native enzyme starts at 330 and ends at 340 nm. At the wavelength of 421 nm the order of the spectra from below becomes the opposite starting with that at 40 min, 35, 30, 25, 20, native, 15, 10, initial [t0] and 5 min.
The presence of the isosbestic points and the time course conversion of the spectra indicates the formation of different oxidation products of the native enzyme and/or the recovery of the enzyme with time especially within the interval between 15 and 20 min where the spectra are the closest to the LPO native enzyme. The products of the enzyme and hydrogen peroxide could be compound I, compound II or compound III. Compound I is transformed into compound II or reduced in the presence of I− into [EOI]− intermediate, which can regenerate the native enzyme36 Compound III is formed when excess H2O2 is present.
Similarly, in the LPO–gua–H2O2 system the time course reconversion of the LPO–guaiacol system is observed (Fig. 6C), where there is a decrease in the absorption of the oxidized form of guaiacol at 470 nm. The lower spectra shown starting from below are for LPO–gua, followed by LPO–gua–H2O2. This was measured after 20 min from the initial record following the addition of H2O2, then 15, 10, 5 min and the initial measurement immediately after adding H2O2. Thus, the native enzyme–gua system will be regenerated after a certain period of adding H2O2. In this way, the kinetic measurements were recorded immediately after the addition of H2O2, before any decomposition of H2O2, or the production of any oxidized substrate product or any regeneration of the initial reactants.
Due to solubility problems, thioamide ligand solutions contained in addition to the buffer 20% DMSO. The amounts of DMSO inside the cell varied between 0.4–4%. To assure that DMSO had no effect on the enzyme a controlled experiment containing the same amounts of DMSO as in the sample with the inhibitors (thioamides) was performed. The control shows no deviation by increasing amounts of DMSO. Thus, the activity of the enzyme in the presence and absence of DMSO was found to be the same throughout the study. The degree of LOX inhibition (A, %) in the presence of the complexes was calculated according to the equation:33

The value of the initial rate (υ0, μM min−1) was calculated according to the formula:

Table 3 gives the value of the IC50 of all these ligands. The IC50 values of LPO enzyme activity inhibition caused by the thioamide ligands lead to their classification into three classes. The first set with the lowest concentration required (<10 μM) constitute the thioamides DTUC, NiBZIM, MBZT and PySH. Low concentrations between 10 and 30 μM are obtained in the case of CMBZT, BZIM, MPmCl, PmSH, MBZIM and MMI. Moderate concentrations between 30 and 80 μM are observed for PTU, TUC, MTU, MNA, PyOH, TU, and MTZT. The large ineffective concentrations (>400 μM) are for PmOH, THP, NMBZT, and PyTU.
The IC50 values vary depending on the type of peroxidase and the substrate used in addition to the conditions of the experiment. IC50 values of 6.6 μM37 and 7 μM38 were reported for MMI using LPO as the peroxidase and 2,2′-azino-bis-3-ethyl-benzothiazoline sulfonic acid (ABTS) as the substrate. In our case the IC50 value of MMI is 28.5 μM which is higher than the reported value, while PTU has its IC50 value equal to 34.30 μM which is lower than the value reported (45 μM)38 with ABTS substrate. The IC = 47.8 μM of MTU is identical to that reported by Roy and Mugesh38 using also ABTS as a substrate.
Conclusions
The anti-thyroidal activity of thioamides is attributed to the following: (a) the ability of the thioamides to form charge transfer complexes with the active iodine. (b) The chemical oxidation of the thioamides by the active iodine causing the formation of the disulfide. (c) Their ability to inactivate the thyroid peroxidase enzyme TPO, the catalyst for the iodination and coupling processes important for the synthesis of the thyroid hormones. (d) The ability of some of the thioamides to inhibit iodothyronine deiodinase (ID-1), an enzyme responsible for the monodeiodination of the T4 prohormone to the active hormone T3, as proposed by du Mont et al.21The kinetic study of the thioamide action performed here on LPO showed that the degree of inhibition ranges from 3.62 μΜ as highest value (DTUC) to 1155.25 μΜ for the lowest (THP) (Table 3). The drugs MMI (28.52 μΜ) and PTU (34.30 μΜ) showed an intermediate inhibition strength as expected since MMI is known to reduce the active diiodine,20 while PTU to inhibit iodothyronine deiodinase (ID-1).3,21–23
The study of the inhibition of the catalytic effect of FeTPPCl (model for the active site of TPO), confirmed the kinetic study results of LPO above. It was found that the strong and medium inhibition occurred with the ligands that are able to form disulfides and ionic compounds (with diiodine and/or iron (Table 2).1–9 NMBZT shows also a relatively high percent of inhibition (20.2%). The crystal structure of NMBZT exists in two forms the spoke structure [LI2]5 and the iodonium salt 1 and also it forms in the presence of iron center an ionic compound of formula {[L2I]+[FeCl4−]},5 hence, its high degree of inhibition could be attributed to its ionic interaction with the iron center of the catalyst FeTPPCl. The weak or no inhibition was observed by the ligands that can form only the spoke structure like PTU and CMBZT.1–9
DFT calculations of the geometric and energetic profiles of the inhibition process revealed that the thermodynamically and kinetically favored reaction pathway for both the quartet and sextet states involves as a first step the coordination of H2O2 to FeTPPCl complex followed by the elimination of H2O to yield [FeCl(TPP)(OI)]− complex, which upon elimination of Cl− affords the intermediate FeTPP(OI) complex. The intermediate [FeCl(TPP)(OI)]− and/or FeTPP(OI) are the crucial species intervening in the inhibition of the catalytic activity of FeTPPCl by thioamides. It was also found that the inhibition mechanism of the active site of TPO involves ionic interactions with the [FeCl(TPP)(OI)]− and/or FeTPP(OI) intermediates. These results agree with the reaction path of Scheme 1.15,19
In conclusion the thioamides DTUC, NiBZIM, MBZT and PySH were found to inhibit the strongest, LPO activity (a model of TPO) with IC50 values <10 μM,(see Table 3).The thioamides MBZT, MMI, PySH, PmSH and MBZIM show a high degree of inhibition (between 27% and 19%) of the catalytic activity of FeTPPCl on the oxidation of iodide anion to I3− by H2O2 (see Table 2).
Thus, thioamides that inhibit LPO and FeTPPCl activities, may interfere in the first stage of thyroide hormone synthesis, which involves the oxidation of I− to active I2.2,21 Other thioamides used as antithyroid drugs may interfere in other stages of the mechanism, as already reported.2,21
Experimental
Materials and instruments
The chemicals were used with no further purification. Diiodine (I2), potassium iodide (KI), N-methyl-2-mercapto-benzothiazole (NMBZT), 2-mercapto-benzothiazole (MBZT), 5-chloro-2-mercapto benzothiazole (CMBZT), 2-mercaptothiazolidine (MTZT), 2-mercaptopyridine (PySH), thiourea (TU), 1,3-bis(3-pyridyl-methyl)-2-thiourea (PyTU), 2-mercapto-3,4,5,6-tetrahydropyrimidine (THP), 2-mercaptopyrimidine (PmSH), 6-propyl-2-thiouracil (PTU), 6-methyl-2-thiouracil (MTU), di-thiouracil-2,4 (DTUC), 2-mercapto-4-methyl-pyrimidine hydro chloride (MPmCl), methimazole (MMI), 2-mercapto-benzimidazole (BZIM), 5-nitro-2-mercapto-benzimidazole (NiMBZIM), 5-methyl-2-mercapto-benzimidazole (MBZIM), 2-hydroxy-pyrimidine (PmOH), 2-hydroxypyridine (PyOH), meso-tetraphenylporphyrin (Fe(TPP)Cl) were purchased from Aldrich, Sigma-Aldrich, Merch-Schunchardt or Ferak. All solvents used were of reagent grade. Mössbauer spectra were recorded with a conventional constant acceleration spectrometer equipped with a 57Co(Rh) source and the parameters were obtained by a least-squares minimization program assuming Lorentzian line shapes. The Mössbauer spectrometer was calibrated with an α-Fe absorber and all isomer shift values reported here are relative to iron at room temperature. Mössbauer spectra were collected at RT and 77 K using a liquid N2 Mössbauer cryostat (Oxford).A Jasco UV/vis/NIR V570 series spectrophotometer was used to obtain electronic absorption spectra.
Synthesis and crystallization of 1
Slow evaporation of the solution resulting from the reaction of di-iodine with N-methyl-2-mercapto-benzothiazole (NMBZT) in the presence of ferric tetra-phenyl-porphyrin chloride (FeTPPCl) in 6∶3∶1 (I2∶thioamide∶FeTPPCl) molar ratio, in dichloromethane gives a mixture of products consisting of the iodonium salt of formula {[(NMBZT)2I]+·[(I5−·I2)n]}n (1) and the FeTPPpCl (2).
Crystal data
Crystals of complexes 1 and 2 suitable for single crystal analysis by X-ray crystallography were grown by slow evaporation of the filtrates from the reaction medium. Data were collected at 293(2) K by the ω scan technique in the θ range 2.5° to 26.5° for 1 and 2° to 30° for 2 on a KUMA KM4CCD four-circle diffractometer39 with a CCD detector, using graphite-monochromated MoKα (λ = 0.71073 Å) radiation. Cell parameters were determined by least-squares fit.40 All data were corrected for Lorentz polarization effects and absorption.40,41 The structures were solved with direct methods with SHELXS9742 and refined by full-matrix least-squares procedures on F2 with SHELXL97.43 All nonhydrogen atoms were refined anisotropically; hydrogen atoms were located at calculated positions and refined as a riding model with isotropic thermal parameters fixed at 1.2 times the Ueq of the carrier atom.1: C16H14I8N2S4: MW = 1377.73, monoclinic, P21/c, a = 9.4596(8) Å, b = 18.2484(15) Å, c = 18.6963(13) Å, β = 98.669(6)°, V = 3190.5(4) Å3, Z = 4, ρ(cald) = 2.87 g cm−3, μ = 8.05 mm−1, reflections collected: 6820, independent: 3261, Rint = 0.1674. Final R indices [I > 2σ(I)]; R1 = 0.087, wR2 = 0.173, S = 1.26, max/min Δρ: 2.21/−1.76 e Å−3.
2: C44H28ClFeN4: MW = 704.00, tetragonal I4, a = 13.563(2) Å, c = 9.8370(12) Å, V = 1809.4(3) Å3, Z = 2, ρ(cald) = 1.29 g cm−3, μ = 0.53 mm−1, reflections collected: 7734, independent: 1777, Rint = 0.031. Final R indices [I > 2σ(I)]; R1 = 0.037, wR2 = 0.091, S = 1.08., max/min Δρ: 0.17/−0.28 e Å−3.
Crystallographic data (excluding structure factors) for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, No. CCDC 783327 (1) and 783326 (2).
Computational details
All calculations were performed in the framework of the DFT implemented in the GAUSSIAN03 package44 using the generalized gradient approximation (GGA) BP86 functional including Becke's 1988 exchange functional (usually abbreviated as B88 or just B)45 and Perdiew's 1986 correlation functional denoted as P86.46 The basis sets employed for the iron and iodine atoms were the Gaussian basis sets of quadruple zeta valence quality (Def2-QZVPP) developed by Weigend and Ahlrichs47 and the 6-31G(d,p) for all other main group elements (E). Full geometry optimization was performed for each structure using Schlegel's analytical gradient method48 and the attainment of the energy minimum was verified by calculating the vibrational frequencies that result in absence of imaginary eigenvalues. This was achieved with the SCF convergence on the density matrix of at least 10−9 and the rms force less than 10−4 au. All bond lengths and bond angles were optimized to better than 0.001 Å and 0.1°, respectively. The computed electronic energies were corrected for zero point energy (ZPE) differences. The natural bond orbital (NBO) population analysis was performed using Weinhold's methodology.49,50 Wiberg Bond Indices (WBI) were calculated using the AOMix program.51,52Acknowledgements
This work was carried out in partial fulfillment of the requirements for a PhD thesis of Dr G.J.C. under the supervision of S.K.H. within the framework of the graduate program in Bioinorganic Chemistry coordinated by N.H. G.J.C. acknowledges the State Scholarship Foundation of the Scholarship for graduate studies.References
- V. Daga, S. K. Hadjikakou, N. Hadjiliadis, M. Kubicki, J. H. Z. dos Santos and I. S. Butler, Eur. J. Inorg. Chem., 2002, 1718–1728 CrossRef CAS.
- C. D. Antoniadis, G. Corban, S. K. Hadjikakou, N. Hadjiliadis, M. Kubicki, S. Warner and I. S. Butler, Eur. J. Inorg. Chem., 2003, 1635–1640 CrossRef CAS.
- C. D. Antoniadis, S. K. Hadjikakou, N. Hadjiliadis, M. Kubicki and I. S. Butler, Eur. J. Inorg. Chem., 2004, 4324–4329 CrossRef CAS.
- C. D. Antoniadis, S. K. Hadjikakou, N. Hadjiliadis, M. Kubicki and I. S. Butler, New J. Chem., 2005, 29, 714–720 RSC.
- G. J. Corban, S. K. Hadjikakou, N. Hadjiliadis, M. Kubicki, E. R. T. Tiekink, I. S. Butler, E. Drougas and A. M. Kosmas, Inorg. Chem., 2005, 44, 8617–8627 CrossRef CAS.
- C. D. Antoniadis, S. K. Hadjikakou, N. Hadjiliadis, A. Papakyriakou, M. Baril and I. S. Butler, Chem.–Eur. J., 2006, 12, 6888–6897 CrossRef CAS.
- S. K. Hadjikakou and N. Hadjiliadis, Bioinorg. Chem. Appl., 2006, 2006, 1–10 Search PubMed , Article ID 60291.
- J. H. Z. dos Santos, I. S. Butler, V. Daga, S. K. Hadjikakou and N. Hadjiliadis, Spectrochim. Acta, Part A, 2002, 58, 2725–2735 CrossRef.
- I.-E. Parigoridi, G. J. Corban, S. K. Hadjikakou, N. Hadjiliadis, N. Kourkoumelis, G. Kostakis, V. Psycharis, C. P. Raptopoulouc and M. Kubicki, Dalton Trans., 2008, 5159–5165 RSC.
- A. Taurog, in Endocrinology, ed. L. DeGroot, Accademic Press, London, 1979, vol. 1, p. 331 Search PubMed.
- D. R. Morris and L. P. Hager, J. Biol. Chem., 1966, 241, 3582–3589 CAS.
- A. Nakataka and H. Hidaka, J. Clin. Endocrinol. Metab., 1976, 43, 152–158 CrossRef.
- Martidale, The Extra Pharmacopoeia, ed. J. E. F. Raynolds, The Pharmaceutical Press, London, 28th edn, 1982 Search PubMed.
- P. J. O'Brien, Chem.-Biol. Interact., 2000, 129, 113–139 CrossRef CAS.
- A. B. Rawitch, A. Taurog, S. B. Chernoff and M. L. Dorris, Arch. Biochem. Biophys., 1979, 194, 244–257 CrossRef CAS.
- R. P. Magnusson, A. Taurog and M. L. Dorris, J. Biol. Chem., 1984, 259, 197–205 CAS.
- R. P. Magnusson, A. Taurog and M. L. Dorris, J. Biol. Chem., 1984, 259, 13783–13790 CAS.
- M. Huwiler, U. Burgi and H. Kohler, Eur. J. Biochem., 1985, 147, 469–476 CAS.
- P. N. Jayaram, G. Roy and G. Mugesh, J. Chem. Sci., 2008, 120, 143–154 CrossRef CAS.
- M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis and G. Verani, J. Am. Chem. Soc., 2002, 124, 4538–4539 CrossRef CAS.
- W. W. du Mont, G. Mugesh, C. Wismach and P. G. Jones, Angew. Chem., Int. Ed., 2001, 40, 2486–2489 CrossRef CAS.
- M. J. Berry, J. D. Kieffer, J. W. Harney and P. R. Larsen, J. Biol. Chem., 1991, 266, 14155–14158 CAS.
- M. J. Berry, L. Banu and P. R. Larsen, Nature, 1991, 349, 438–440 CrossRef CAS.
- F. Demartin, P. Deplano, F. A. Devillanova, F. Isaia, V. Lippolis and G. Veranil, Inorg. Chem., 1993, 32, 3694–3699 CrossRef CAS.
- E. B. Fleischer, C. K. Miller and L. E. Webb, J. Am. Chem. Soc., 1964, 86, 2342 CrossRef.
- J. L. Hoard, G. H. Cohen and M. D. Glick, J. Am. Chem. Soc., 1967, 89, 1992–1996 CrossRef CAS.
- H. Jenzer, U. Burgi and H. Kohler, Biochem. Biophys. Res. Commun., 1987, 142, 552–558 CrossRef CAS.
- M. Huwiler and H. Kohler, Eur. J. Biochem., 1984, 141, 69–74 CrossRef CAS.
- D. Deme, E. Fimiani, J. Pommier and J. Nunez, Eur. J. Biochem., 1975, 51, 329–336 CrossRef CAS.
- G. Traylor, W. A. Lee and D. V. Stynes, J. Am. Chem. Soc., 1984, 106, 755–764 CrossRef CAS.
- M. Huwiler, U. Burgi and H. Kohler, Eur. J. Biochem., 1985, 147, 469–476 CAS.
- A. Decker and E. I. Solomon, Angew. Chem., Int. Ed., 2005, 44, 2252 CrossRef CAS.
- E. O. de Aspuru and A. M. L. Zaton, Spectrochim. Acta, Part A, 1999, 55, 2343–2346 CrossRef.
- J. Nunez, Methods Enzymol., 1984, 107, 477–488 Search PubMed.
- R. E. Huber, L. A. Edwards and T. J. Carne, J. Biol. Chem., 1989, 264, 1381–1386 CAS.
- H. Kohler, A. Taurog and H. B. Dunford, Arch. Biochem. Biophys., 1988, 264, 438–649 CrossRef CAS.
- G. Roy, M. Nethaji and G. Mugesh, J. Am. Chem. Soc., 2004, 126, 2712–2713 CrossRef CAS.
- G. Roy and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 15207–15217 CrossRef CAS.
- KUMA KM-4CCD User Manual, KUMA Diffraction, Wroclaw, Poland, 1999 Search PubMed.
- CrysAlis, Program for Reduction of the Data from KUMA CCD Diffractometer, KUMA Diffraction, Wroclaw, Poland, 1999 Search PubMed.
- R. H. Blessing, J. Appl. Crystallogr., 1989, 22, 396 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97 Program for refinement of crystal structures, University of Gottingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- F. Weinhold, in The Encyclopedia of Computational Chemistry, ed. P. v. R. Schleyer, John Wiley & Sons, Chichester, 1998, p. 1792 Search PubMed.
- S. I. Gorelsky, AOMixProgram for Molecular Orbital Analysis York University, Toronto, Canada(http://www.sg-chem.net) Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: (a) The most relevant Frontier Molecular Orbitals; (b) equilibrium geometries of all stationary points located on the quartet and sextet potential energy surfaces of the inhibition of the catalytic activity of FeTppCl by the thioamide, (c) 3D isospin surfaces of total spin density of the stationary points and sextet potential energy surfaces of the inhibition of the catalytic activity of FeTppCl by the thioamide compounds, (d) cartesian coordinates and energy data of all structures studied. For ESI see DOI: 10.1039/c0nj00626b |
| ‡ Present address: Inorganic Chemistry Laboratory, Department of Chemistry, College of Science, UAE University, 17551, Al Ain, United Arab Emirates. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011 |