Far red and NIR dye-peptoid conjugates for efficient immune cell labelling and tracking in preclinical models†
Kevin
Dhaliwal
*a,
Géraldine
Escher
b,
Asier
Unciti-Broceta
c,
Neil
McDonald
a,
A. John
Simpson
d,
Chris
Haslett
a and
Mark
Bradley
*b
aMRC Centre for Inflammation Research, University of Edinburgh, 47 Little France Crescent, Edinburgh, UK. E-mail: kdhaliwa@staffmail.ed.ac.uk
bSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, UK. E-mail: mark.bradley@ed.ac.uk
cEdinburgh Cancer Research Centre, MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, Crewe Road South, Edinburgh, UK
dInstitute of Cellular Medicine, Medical School, Newcastle University, Newcastle upon Tyne, UK
First published on 30th August 2011
Abstract
Innate immune cell ingress into the site of inflammation is central for the efficient clearance of pathogens. However, in some circumstances the inflammatory response may become pathogenic to the host. Understanding the temporal ingress of innate immune cells is thus essential to predict both the defensive and adverse effects mediated by these cells in an infectious process and for developing novel therapeutic strategies. In this context, optical imaging has emerged as a powerful technique for the visualization of specific cellular events in preclinical models. Herein we describe a non-covalent tagging strategy to stably label innate immune cells using in vivo-traceable cell penetrating peptoids and their application in real-time imaging of cell migration in murine models, thereby providing a sensitive and cost effective way to visualize cellular recruitment in preclinical models of inflammation.
Introduction
Phagocytic cells such as monocytes, macrophages and neutrophils are white blood cells responsible for the host innate defense against infection.1 Circulating monocytes differentiate into macrophages which are resident phagocytes involved in the orchestration of the innate response upon local pathogen recognition. Neutrophils are circulating granulocytes recruited to sites of acute inflammation and constitute the first line of cellular defense in bacterial infection.2 However, although neutrophils are essential for the efficient clearance of intracellular and extracellular microbial pathogens, they also have the potential to cause tissue damage as a result of a dynamic imbalance between the levels of proteases and protective protease modulators.3 Given the potential of neutrophils to injure tissues, the inflammatory response has evolved to facilitate their efficient removal after bacterial clearance. However, in some instances neutrophils may continue to be recruited and in the face of their poor clearance toxic neutrophil derived “products” may perpetuate the inflammatory response.3,4 Understanding the migration and trafficking patterns of phagocytes is thus central to predict and control both the defensive and adverse effects mediated by these cells as well as to potentially devise novel immune cell-delivered therapeutic strategies.5Optical imaging is widely used in preclinical research offering a number of practical advantages compared to other imaging modalities, specifically its sensitivity, moderate cost, non-invasiveness and direct cellular readout.6,7 Among the possible chemical and biological strategies that can be employed to “optically” label primary cells (e.g.antibody labelling, genetic labelling – with bioluminescent or fluorescent GFP-like proteins–, and chemical labelling),7–9 synthetic probes are probably the most cost-effective and straightforward, just requiring cell isolation, labelling with the probe of choice and subsequent infusion back into the host.9,10 Nevertheless, due to the high sensitivity to inflammatory stimulation and short lifetime of immune cells,11probe labelling needs to have a number of essential features namely the ability to label cells rapidly (within minutes), lack of toxicity or alteration of normal cell behaviour, and be stably retained within the cells for long duration studies (days to weeks).
Currently four types of chemically-based fluorescent probes are used for in vivocell tracking studies: (i) cell-permeable dyes which enter cells by difussion and bind strongly to intracellular structures by non-covalent interactions (e.g.DNA-binding dyes, membrane-inserting fluorescent dyes, etc);12 (ii) labelling probes that covalently link to intracellular components,13,14 with dyes such as 5-chloromethylfluorescein diacetate (also known as CellTracker Green) which has demonstrated high efficacy and delays the “label leaking” from the cells; and (iii) dye-labelled cell-penetrating probes, which enter cells by probe-mediated translocation and do not bind permanently to intracellular structures (e.g. fluorescently-labelled cell-penetrating peptides (CPPs)),15 and (iv) cell parenting micro and nanoparticles.16 The first two kinds of labelling probes are particularly effective and have been widely used in vivo.12–14 However, high levels of labelling equates to extensive protein or DNA modification which is undesirable as it may affect cell viability and normal cell behaviour. This risk can be circumvented by using reagents of the third category, fluorescently-labelled cell penetrating peptides, which are capable of translocating a wide range of covalently-linked cargoes across the plasma membrane.17 However these are often susceptible to protease degradation, can be toxic and display lower labelling efficiency for primary cells.18
An attractive strategy to address these issues is the use of cell penetrating peptoids,19–22 which are known to be resistant to proteolysis and have been shown by our group to offer some of the highest cell labelling/delivery efficacies known.23 In particular the nona(N-aminohexyl)peptoid (9-mer) which display remarkable delivery properties, being able to efficiently label different cell lines within minutes (Fig. 1).23 In this study we report the synthesis of peptoids conjugated to far red to near infra-red (NIR) dyes and their efficient application for ex vivolabelling and in vivo real-time tracking of primary phagocytes.
 | ||
| Fig. 1 Cell-penetrating peptoid.23 | ||
Results and discussion
Fluorescently-labelled cell penetrating peptoids were synthesized using microwave-assisted solid-phase synthesis as shown in Scheme 1. Peptoids were assembled following an Fmoc-based strategy on Rink amide-functionalized aminomethyl polystyrene resin 1 using orthogonally-protected lysine-like monomer 2.24,25 An in-house-optimized monomer-based synthesis was used23,25 instead of the submonomer approach,26 driven by the length of the desired product (9-mer) and the highly hydrophilic nature of the final conjugates that make purification of truncations problematic. Monomer assemblings were carried out using a two-step procedure: (i) piperidine-mediated Fmoc deprotection; (ii) followed by coupling with monomer 2 using HOBt/DIC. This procedure was repeated nine times to obtain the oligomer with desired length. The resulting resin 3 was Fmoc deprotected and, using the same procedure, coupled with a six carbon spacer (Fmoc-Ahx-OH) to give resin 4 (this spacer separates the dye from the peptoid and allows efficient conjugation of the dye). Finally, dyes were coupled onto the resin-bound peptoid 4 which was subsequently, Boc-deprotected and cleaved from the resin under acidic conditions to give 5 and 6. | ||
Scheme 1
Fmoc solid-phase synthesis of fluorescently-labelled cell penetrating peptoids. Reagents and conditions: (i) 20% piperidine in DMF, 10 min (×2); (ii) monomer 2 (3 equiv.), DIC (3 equiv.), HOBt (3 equiv.), DMF, μW, 60 °C, 20 min; (iii) Fmoc-Ahx-OH (3 equiv.), DIC (3 equiv.), HOBt (3 equiv.), DMF, μW, 60 °C, 20 min; (iv) activated dye (see ESI), DMF; (v) TFA/TIS/H2O (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5), 4 h. :2.5:2.5), 4 h. | ||
The cyanine dyes are a group of fluorophores commonly used for optical imaging applications due to their tunable wavelengths (red to near infrared) and high extinction coefficients.27 One of the most attractive subgroups are the indocyanine dyes that, in contrast with other cyanines dyes, avoid self-aggregation.28 The polymethine indocyanine dyes (Cy) used to label the peptoid were readily generated by minor modifications to a generic synthetic procedure to allow conjugation to the peptoid.29 As expected, dye-labelled peptoids 5 and 6 displayed emission maximum peaks in the far red (710 nm) and NIR (779 nm) region respectively (see Fig. 2a and ESI).
 | ||
| Fig. 2 (a) Structures of the dye-labelled peptoids 5 and 6. (b) Comparative fluorescence intensity of labelled primary murine monocytes following 10 min incubation with Cy7-9-mer 6 at various concentrations as measured by flow cytometry. (c–d) Confocal images of (c) monocytes and (d) neutrophils after 10 min incubation with 5μM Cy5.5-9-mer 5. Left: Cy5.5-9-mer 5 (633 nm excitation). Middle: DAPI nuclei staining (405 nm excitation). Right: composite image. Scale bar: 2 μm. | ||
Assessment of the ex vivolabelling abilities of 5 and 6 was carried out with innate immune cells. Primary murine bone marrow monocytes and neutrophils were retrieved, incubated with dye-labelled peptoids 5 and 6 for 10 min and analyzed by flow cytometry and confocal microscopy. Flow cytometry analyses showed 100% cell labelling, with a dose-dependent increase in fluorescence intensity (Fig. 2b) and no signs of cytotoxicity by MTT or TO-PRO-3 exclusion assays (see ESI). The cytoplasmic location of the labelled-peptoids was demonstrated by confocal microscopy (Fig. 2c, d), whilst inflammatory cytokines liberated from primary murine monocytes and neutrophils treated with Cy7-9-mer 6 were not elevated over controls.
The spectral properties of the dyes were optimal for live whole body, non-invasive biophotonic imaging in mice,30 allowing investigation of dynamic migration and functional viability of labelled phagocytes which were infused intravenously into syngeneic mice that had localized subcutaneous inflammation.31 As shown in Fig. 3a, infused primary monocytes loaded with Cy7-9-mer 6 demonstrated tracking to sites of subcutaneous inflammation.
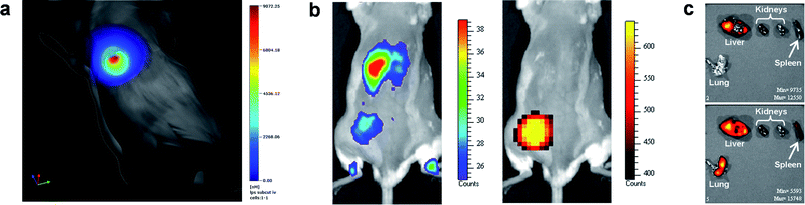 | ||
| Fig. 3 Non-invasive optical imaging of mice with localized inflammation. (a) Tracking of Cy7-9-mer 6 labelled primary monocytes to LPS-induced subcutaneous inflammation using thoracic gated tomographic imaging. (b) Tracking of Cy5.5-9-mer 5 labelled primary neutrophils to subcutaneous inflammation induced by LPS-impregnated matrigel implanted in the left thigh of a mouse. Right panel: luminescence in vivo imaging after intraperitoneal administration of luminol showing myeloperoxidase activity in the site of inflammation. Left panel: fluorescence in vivo imaging of Cy5.5-9-mer 5 labelled neutrophils after intravenous infusion showing unilateral recruitment to the site of injury in the same mouse. (c) Dual channel fluorescent imaging of neutrophil recruitment to different organs after LPS-induced lung injury. Cy5.5-9-mer 5 labelled neutrophils were adoptively transferred 4 h after injury and Cy7-9-mer 6 labelled neutrophils 24 h after injury. 48 h after LPS lung injury, organs were removed and imaged under different channels (upper panel: ex/em 680/720 nm; lower panel: ex/em 745/820 nm), showing Cy5.5-9-mer 5 labelled neutrophil presence exclusively in liver while Cy7-9-mer 6 labelled neutrophils were present in liver, spleen and lungs of the same animal. | ||
Further evidence for selective homing of innate immune cells was carried out utilising a recently described luminescent model32 for imaging sites of inflamation based on the chemiluminescent oxidation of luminol due to reactive oxygen species produced by constitutive neutrophil myeloperoxidase. This model was used to identify an adoptive neutrophil-specific myeloperoxidase activity in a model of subcutaneous inflammation induced by lipopolysaccharide (LPS)-impregnated matrigel implanted in the left thigh of the mice. Cy5.5-9-mer 5 labelled neutrophils were infused intravenously in the injured mice and, after 4 h, luminol administered intraperitoneally. Optical imaging showed selective unilateral accumulation of Cy5.5-9-mer labelled neutrophils within the site of active inflammation with signal colocalisation with myeloperoxidase-mediated bioluminiscence of luminol (Fig. 3b).
The ability to spectrally unmix the far red and near infrared dyes and facilitate dual fluorescent channel imaging was exploited to delineate kinetic recruitment of adoptively transferred neutrophils to sites of inflammation. Two neutrophil populations were separately labelled with Cy5.5-9-mer 5 and Cy7-9-mer 6 and then infused intravenously at different time points after injury: 4h and 24 h after inducing lung injury, respectively. As shown in Fig. 3c, imaging of retrieved organs showed distinct time-dependent migration pattern, with only neutrophils infused 24 h after injury being efficiently recruited to the site of inflammation (lung).
Neutrophils and monocytes are primary cells recruited at the earliest stages of inflammation and their tracking has previously required the utilisation of radioisotopic methods due to difficulties in their rapid and non-toxic labelling.33 Our data using optical agents is the first description of tracking monocytes and neutrophilsin vivo utilising internalised non-covalent optical tracers as opposed to membrane dyes. Optimal cell isolation, labelling and injection into syngeneic mice was performed within 2 h of marrow harvest, with a total peptoid-cell incubation time of ten minutes. This speed of labelling being specifically facilitated by the nona(N-aminohexyl)peptoid (Fig. 1).
Finally, the in situ functional viability of peptoid-treated cells was demonstrated by labelling primary murine macrophages with Cy7-9-mer 6 and simultaneously transfecting with a replication-deficient adenovirus encoding luciferase. These cells were instilled directly into the murine lung and imaged 24 h later. Fluorescence imaging confirmed localisation in the lung, while intraperitoneal luciferin injection led to the generation of luminescence in the same region (Fig. 4), thus demonstrating the viability of labelled, adoptively transferred cells.
 | ||
| Fig. 4 In vivo functional viability study. Primary murine macrophages were chemically labelled with Cy7-9-mer 6 and labelled by transduction with an adenovirus encoding luciferase. These doubly-labelled macrophages were instilled into the murine lung and imaged 24 h later. Left panel shows fluorescence imaging under a Cy7 filter (ex/em 745/820 nm). Right panel shows luminescence imaging of the same animal after intraperitoneal luciferin administration. Images captured on a Xenogen Spectrum. | ||
Conclusions
In summary, we have developed an efficient method to track innate immune cell migrationin vivo using far red and NIR dye-labelled peptoids. Thanks to their proteolytic resistance and high labelling efficiency, neutrophils and monocytes were tracked for over three days, allowing the visualization of their recruitment to sites of inflammation. Their remarkably fast labelling, low toxicity and long-term stability, make the use of fluorescently-labelled peptoids highly attractive tools for in vivo optical imaging.The authors would like to acknowledge funding from the EC FP6 (Marie Curie EST Fellowship to G.E.) and the MRC (K.D, C.H and M.B.). We also thank the MRC IGMM (A.U.B.) and Jules Thorne Trust (A.J.S.).
Notes and references
- M. T. Silva, J. Leukocyte Biol., 2009, 87, 93–106 Search PubMed.
- T. S. Wilkinson, K. Dhaliwal, T. W. Hamilton, A. F. Lipka, L. Farrell, D. J. Davidson, R. Duffin, A. C. Morris, C. Haslett, J. R. W. Govan, C. D. Gregory, J.-M. Sallenave and A. J. Simpson, Am. J. Pathol., 2009, 174, 1338–1346 CrossRef CAS.
- A. G. Rossi, J. M. Hallett, D. A. Sawatzky, M. M. Teixeira and C. Haslett, Biochem. Soc. Trans., 2007, 35, 288–291 CrossRef CAS.
- S. Mandl, C. Schimmelpfennig, M. Edinger, R. S. Negrin and C. H. Contag, J. Cell. Biochem., 2002, 87, 239–248 CrossRef.
- C. Haslett, Am. J. Respir. Crit. Care Med., 1999, 160, S5–11 Search PubMed.
- V. Ntziachristos, J. Ripoll, L. V. Wang and R. Weissleder, Nat. Biotechnol., 2005, 23, 313–320 CrossRef CAS.
- B. Isherwood, P. Timpson, E. J. McGhee, K. I. Anderson, M. Canel, A. Serrels, V. G. Brunton and N. O. Carragher, Pharmaceutics, 2011, 3, 141–170 Search PubMed.
- H. Hong, Y. Yang, Y. Zhang and Weibo Cai, Curr. Top. Med. Chem., 2010, 10, 1237–1248 CrossRef CAS.
- K. M. Marks and G. P. Nolan, Nat. Methods, 2006, 3, 591–596 CrossRef CAS.
- K. Dhaliwal, L. Alexander, G. Escher, A. Unciti-Broceta, M. Jansen, N. Mcdonald, J. M. Cardenas-Maestre, R. Sanchez-Martin, J. Simpson, C. Haslett and M. Bradley, Faraday Discuss., 2011, 149, 107–114 RSC.
- E. L. Becker, J. Leukocyte Biol., 1990, 47, 378–89 Search PubMed.
- C. R. Parish, Immunol. Cell Biol., 1999, 77, 499–508 CrossRef CAS.
- F. K. Swirski, C. R. Berger, J.-L. Figueiredo, T. R. Mempel, U. H.v. Andrian, M. J. Pittet and R. Weissleder, PLoS One, 2007, 2, e1075 CrossRef.
- A. E. Foster, S. Kwon, S. Ke, A. Lu, K. Eldin, E. Sevick-Muraca and C. M. Rooney, Appl. Opt., 2008, 47, 5944–5952 CrossRef CAS.
- G. Tünnemann and M. C. Cardoso, in Membrane-Active peptides: methods and results on structure and function, ed. M. Castanho, IUL Publishers, La Jolla (US), pp. 331–362 Search PubMed.
- R. M. Sachez-Martin, M. Muzerelle, N. Chitkul, S. E. How, S. Mittoo and M. Bradley, ChemBioChem, 2005, 6, 1341–1345 CrossRef CAS.
- J. S. Wadia and S. F. Dowdy, Curr. Opin. Biotechnol., 2002, 13, 52–56 CrossRef CAS.
- E. Koren, A. Apte, R. R. Sawant, J. Grunwald and V. P. Torchilin, Drug Delivery, 2011, 18, 377–384 CrossRef CAS.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS.
- I. Peretto, R. M. Sanchez-Martin, X. H. Wang, J. Ellard, S. Mittoo and M. Bradley, Chem. Commun., 2003, 2312–2313 RSC.
- R. N. Zuckermann and T. Kodadek, Curr. Opin. Mol Ther., 2009, 11, 299–307 Search PubMed.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282–5335 Search PubMed.
- A. Unciti-Broceta, F. Diezmann, C. Y. Ou-Yang, M. A. Fara and M. Bradley, Bioorg. Med. Chem., 2009, 17, 959–966 CrossRef CAS.
- D. Orain, J. Ellard and M. Bradley, Protecting groups in solid-phase organic synthesis, J. Comb. Chem., 2002, 4, 1–16 CrossRef CAS.
- M. A. Fara, J. J. Diaz-Mochon and M. Bradley, Tetrahedron Lett., 2006, 47, 1011–1014 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef.
- M. Lopalco, E. N. Koini, J. K. Cho and M. Bradley, Org. Biomol. Chem., 2009, 7, 856–859 RSC.
- V. Ntziachristos, C. Bremer and R. Weissleder, Eur. Radiol., 2003, 13, 195–208.
- Animal experimentation was approved by UK regulatory authorities (Project licence 60/3545 from UK Home Office).
- S. Gross, S. T. Gammon, B. L. Moss, D. Rauch, J. Harding, J. W. Heinecke, L. Ratner and D. Piwnica-Worms, Nat. Med., 2009, 15, 455–461 CrossRef CAS.
- F. K. Swirski, M. J. Pittet, M. F. Kircher, E. Aikawa, F. A. Jaffer, P. Libby and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10340–10345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental description of the synthesis of compounds 5 and 6, their characterization, and biological assays. See DOI: 10.1039/c1md00171j |
| This journal is © The Royal Society of Chemistry 2011 |