Discovery of CP-866,087, a mu opioid receptor antagonist for the treatment of alcohol abuse and dependence†
Stanton F.
McHardy‡
*a,
Steven D.
Heck
a,
Sara
Guediche
a,
Monica
Kalman
a,
Martin P.
Allen
a,
Meihua
Tu
a,
Dianne K.
Bryce
b,
Anne W.
Schmidt
b,
Michelle
Vanase-Frawley
b,
Ernesto
Callegari
c,
Shawn
Doran
c,
Nicholas J.
Grahame
d,
Stafford
McLean
b and
Spiros
Liras
*a
aWorldwide Medicinal Chemistry, Pfizer Worldwide Research and Development, Eastern Point Road, Groton, CT 06340, USA. E-mail: spiros.liras@pfizer.com
bNeuroscience Biology Pfizer Worldwide Research and Development, Eastern Point Road, Groton, CT 06340, USA
cPharmacokinetics, Dynamics and Metabolism, Pfizer Worldwide Research and Development, Eastern Point Road, Groton, CT 06340, USA
dDepartment of Psychiatry, Indiana University School of Medicine, Indianapolis, IN 46202, USA
First published on 25th August 2011
Abstract
CP-866,087 (compound 15) is a novel, potent and selective mu opioid receptor antagonist. The design rationale, synthesis, in vitro and in vivo biological evaluation are reported herein. Preclinical efficacy data in disease relevant models measuring reduction of alcohol intake are presented, along with preclinical pharmacokinetic properties which supported the selection of the title compound for clinical evaluation.
Introduction
Non-selective opioid antagonists, such as naltrexone, naloxone and nalmafene have demonstrated clinical efficacy in reduction of ethanol intake.1 The association of opioid receptor antagonism and reduction in ethanol consumption has been studied extensively. It has been postulated that ethanol stimulates the endogenous opioid pathways that in turn activate the dopaminergic signaling of the reward system in the brain.2 Opioid antagonism interferes with dopaminergic signaling and activation of the reward system. While clinical efficacy has been demonstrated with non-selective opioid antagonists, it has not been determined which receptor subtype is the primary driver for efficacy. On this basis, we set out to design and synthesize mu receptor selective antagonists and investigate their potential utility as therapeutics for alcohol abuse and dependence. Mu opioid receptors are located on dopaminargic neurons.3 Stimulation of mu receptors augments dopamine function in the CNS and this is associated with activation of the reward system. Kappa opioid receptors are also found on dopaminargic neurons. However, activation of kappa receptors is thought to inhibit DA function.4 Thus, our research objective was to generate a novel and selective mu opioid receptor antagonist with sufficient pharmacokinetic and disposition characteristics to provide an once a day therapy for alcohol abuse and dependence.Results and discussion
Zimmerman, Caroll et al.5 have shown that 4-phenyl piperidines exhibit potent affinity for the mu receptor. Based on extensive studies on 4-phenyl piperidines it was determined that ring systems which display the aromatic ring in an equatorial conformation deliver mu receptor antagonists (1, Fig. 1). With this knowledge, Pfizer coworkers determined that the novel 3.1.0 bridged bicycle 2, shown in Fig. 1, was a privileged template which displayed the aromatic ring in a locked, equatorial-like configuration. Extensive SAR work suggested that the scaffold delivered potent mu receptor antagonists.6 Given our interest in mu opioid antagonists, we were attracted to this scaffold for several reasons including molecular symmetry, rich SAR which suggested tolerance with amine substitutions and the demonstrated ability to replace phenols with a methyl sulfonamide surrogate.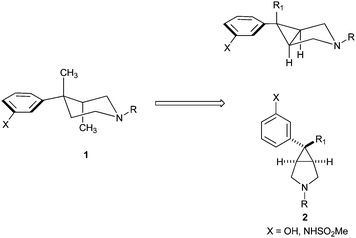 | ||
| Fig. 1 4-Phenyl piperidine derived mu opioid receptor antagonists. | ||
Thus, our first objective was to develop a scalable synthesis which would enable the expanded parallel investigation of two points of diversity; (a) N-substitutions and (b) phenol surrogates. The synthesis plan and its reduction to practice are summarized in Schemes 1 and 2. The synthesis commenced with the conversion of 3-bromophenyl-propane-1-one to the corresponding hydrazone 3 with hydrazine in water-methanol at reflux. The crude hydrazone was treated with MnO2 in dioxane and N-benzyl-pyrrole at room temperature to afford compound 6, via the intermediate diastereomeric cycloaddition products 4 and 5.7 Compound 6 was reduced in high yield with BF3 Et2O and NaBH4 in THF to the desired 3.1.0 bicylic amine 7.
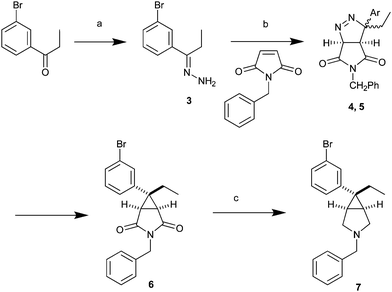 | ||
| Scheme 1 Reagents and conditions: (a) Hydrazine hydrate, MeOH, reflux, 95%; (b) MnO2, dioxane, 1-benzyl-1H-pyrrole-2,5-dione, 35–40% overall from 3; (c) BF3-Et2O, NaBH4, THF, piperazine, H2O, reflux, 95%. | ||
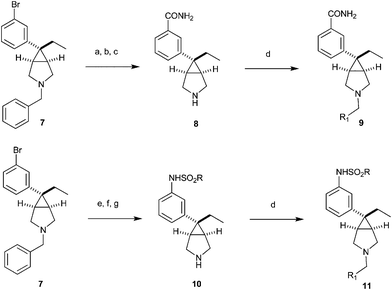 | ||
| Scheme 2 Reagents and conditions: (a) Zn(CN)2, Pd(PPh3)4, 80%; (b) H2O2, K2CO3, DMSO, 60%; (c) Pd/C, MeOH, NH4CO2H, 80%; (d) R1CHO, NaBH(OAc)3, (CH2)2Cl2, 70–90%; (e) Pd(OAc)2, BINAP, benzophenone imine, NaOtBu, HCl,; (f) RSO2Cl, pyridine, 70% for e and f; (g) H2, Pd/C, MeOH, NH4CO2H, 78%. | ||
Amine 7 was further elaborated to the corresponding carboxamidevia a two step sequence which involved first conversion to a nitrile with Zn(CN)2 and Pd(PPh3)4 and subsequent oxidation with H2O2, K2CO3 in DMSO.8,9Debenzylation proceeded smoothly to yield secondary amine 8 which was readily elaborated to 9 to investigate SAR with N-substitutions and the carboxamide as a phenol surrogate. Similarly, amine 7 was converted to a methyl sulfonamidevia transformation of the bromophenyl moiety to an aniline with Pd (OAc)2, BINAP, benzophenone imine10 followed by acid hydrolysis and subsequent treatment with MeSO2Cl and pyridine. Secondary amine 10 was afforded after standard debenzylation conditions. Amine 10 was similarly elaborated to tertiary amine 11 to investigate SAR with N-substitutions and the methyl sulfonamide as a phenol surrogate.
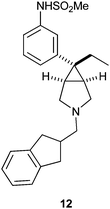
As has been shown in the opioid literature, with phenyl piperidines and aryl morphans, various amine tethers are well tolerated and yield potent mu receptor ligands with variable degrees of selectivity.5 The same was realized with amine tethers for 9 and 11.6 From the early SAR efforts we identified compound 12 as a promising lead and this compound was characterized in depth. Compound 12 exhibited appreciable affinity for the mu receptor and good selectivity over the delta and kappa receptors (Ki of 5 nM, 112 nM, and 56 nM respectively).11,12 Compound 12 resides in lipophilic physicochemical space with a cLogP of 4.1 and TPSA of 58. Based on its attractive in vitro profile, compound 12 was evaluated in key ADME and safety pharmacology screens. In human liver microsomes the compound showed an intrinsic clearance of 19 ml min−1/kg. As is characteristic of numerous lipophilic amines the compound showed considerable metabolism by polymorphic CYP-2D6 (35% enzyme inhibition at 3 μM).13,14 Compound 12 was predicted to be a modest P-gp efflux substrate based on MDR1 transfected MDCK cells data (BA/AB of 3.5).15 Additionally, the compound showed appreciable affinity in a 3H-dofetilide binding assay which is used as a surrogate for predicting interaction with the HERG channel (IC50 of 326 nM).16 Consequently, our attention was focused on devising a uniform strategy to further optimize ADME properties, reduce interaction with CYP-2D6, P-gp and reduce affinity in the 3H-dofetilide binding assay (Table 1).
|
Mu Ki (nM) | 5 |
| Delta Ki (nM) | 112 | |
| Kappa Ki (nM) | 56 | |
| Ratio μ/δ/κ | 1/22/11 | |
| cLogP | 4.1 | |
| TPSA | 58 | |
| Dofetilide IC50 (nM) | 326 | |
| HLM (ml min−1 kg−1) | 19 | |
| CYP-2D6% inh @ 3 μM | 35 | |
| MDR BA/AB | 3.5 |
We determined that the key design objective was reduction of lipophilicity. As demonstrated with related systems, reduction of lipophilicity leads to improvements in microsomal stability.17 Furthermore, we hypothesized that the observed dofetilide affinity was an outcome of pharmacological promiscuity due to lipophilicity.18 With respect to CYP-2D6 interaction, it has been postulated that CYP-2D6 induced oxidation of basic molecules occurs at a site proximal to basic nitrogen, within 5–7 Angstroms.19 Thus, we targeted to incorporate hydroxyl-substituted aliphatic side chains to minimize turnover by CYP-2D6. The incorporation of these tethers was predicted to reduce lipophilicity which we expected would reduce affinity for dofetilide and benefit microsomal stability. Thus, we envisaged that the incorporation of hydroxylgroups on the amine alkyl tethers would serve as a uniform strategy to address all limitations with compound 12. We aimed to incorporate hydroxyl-substituted side chains which would preserve the molecular symmetry inherent to the scaffold and which would present reasonable steric hindrance to reduce the risk for secondary metabolism. Compounds 13, 14 and 15 all meet the design criteria (Fig. 2).
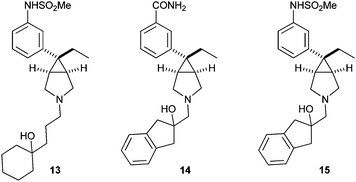 | ||
| Fig. 2 Amines 13, 14 and 15. | ||
Amine 13 was prepared with standard reductive alkylation conditions as reported in Scheme 2. Amines 14 and 15 were accessed as described in Scheme 3. 1H-Inden-2(3H)-one was treated with trimethylsilyl methyl lithium and CeCl3 to afford hydroxy silane 16, which upon treatment with HF in acetonitrile and subsequent oxidation of the ensuing olefin with mCPBA yielded epoxide 17.20 Compound 17 was then reacted with amines 8 and 10 to yield 14 and 15 in good yields.
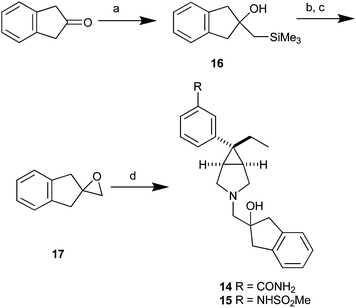 | ||
| Scheme 3 Reagents and conditions: (a) trimethylsilyl methyl lithium, CeCl3, TMEDA 50%; (b) 50% aq HF, CH3CN; (c) mCPBA, NaHCO3, CH2Cl2, 81% over 2 steps; (d) amines 8 and 10, Et3N, EtOH reflux, 60–80%. | ||
Compound 13 showed excellent affinity for the mu receptor (2 nM), modest selectivity over the kappa receptor (16 nM) and excellent selectivity over the delta receptor (166 nM). Relative to 12, compound 13 resides in less lipophilic space with a cLogP of 3.2 and a TPSA of 78. The increased polarity, as desired, yielded an increase in microsomal stability as measured by human liver microsomes (Clint of 9.3 ml min−1 kg−1). In addition, dofetilide activity was reduced (IC50 of 710 nM). As designed, the hydroxylation delivered a compound with reduced interaction with CYP-2D6 (14% inhibition at 3 μM). Similarly, compound 14 exhibited high affinity for the mu receptor (3.5 nM), suggesting the suitability of the primary hydroxamide as a phenol surrogate. The compound retained good to excellent selectivity over the kappa and delta receptors (60 and 108 nM respectively). Compound 14 also resides in less lipophilic space than the predecessor 12 (cLogP of 2.8, TPSA of 67). Consistent with these properties the compound exhibited reduced interaction with CyP-2D6 (17% inhibition at 3 μM), improved stability in human liver microsomes (Clint of 11 ml min−1 kg−1) and reduced activity in dofetilide (IC50 of 2.2 μM), all relative to amine 12. Compound 15 exhibited the best affinity and selectivity profile for the mu, kappa and delta receptors (1, 42, 170 nM respectively). With a cLogP of 3.1 and a TPSA of 78 the compound resides in acceptable physicochemical space. Compound 15 showed reduced interaction with CYP-2D6 (6% inhibition at 3 μM) and improved stability in human liver microsomes relative to 12 (Clint of 12 ml min−1 kg−1). Additionally, amine 15 displayed reduced affinity in 3H-dofetilide relative to 12 (IC50 of 1.6 μM). All compounds were characterized as mu antagonists based on a GTP-γS assay (Table 2). Similarly, all compounds were found to be delta and kappa receptor antagonists in the corresponding GTP-γS assays.
| Compound | 13 | 14 | 15 |
|---|---|---|---|
| Mu Ki (nM) | 2.0 | 3.5 | 1.0 |
| Delta Ki (nM) | 166 | 108 | 170 |
| Kappa Ki (nM) | 16 | 60 | 42 |
| Ratio μ/δ/κ | 1/83/8 | 1/31/17 | 1/170/42 |
| Mu antag. GTP-γS Ki (nM) | 0.18 | 0.16 | 0.28 |
| cLogP | 3.3 | 2.8 | 3.1 |
| TPSA | 78 | 67 | 78 |
| Dofetilide IC50 (nM) | 710 | 2200 | 1600 |
| HLM (ml min−1 kg−1) | 9 | 14 | 12 |
| CYP-2D6% inh @ 3 μM | 14 | 17 | 6 |
| MDR BA/AB | 6.5 | 2.9 | 1.2 |
Compounds 13 and 15 were selected for additional in vivo evaluation based on their overall in vitro profile. These compounds are differentiated in one important dimension which merited detailed investigation. Based on the in vitroMDR data compound 13 was predicted to be an efflux substrate with a BA/AB ratio of 6.5 and compound 15 was predicted not to be an efflux substrate (BA/AB ratio of 1.2). While the compounds are structurally similar, they differentiate in one important physicochemical parameter (amine pKa). Based on calculations compound 15 is predicted to be less basic than compound 13 (pKa of 8.9 and 11.1 respectively).21 Compound 15 forms a strong intra-molecular hydrogen bond in its protonated form, which is likely to contribute to its relatively lower pKa. This was an important design feature. We hypothesized that this parameter was likely to affect interaction with P-gp and thus produce different outcomes with respect to plasma and CSF concentrations required for the desired in vivo efficacy.
To assess in vivo CNS activity both compounds 13 and 15 were evaluated in a rodent tail flick assay. In this assay a focused light source was applied to the tail of a mouse or a rat. The latency to remove the tail from this moderately painful stimulus was measured. In rodents, morphine increases this latency. The increase in latency is interpreted as morphine-induced antinociception which is thought to be mediated by mu receptor agonism. Pretreatment with 13 antagonized the effect of morphine with an ED50 of 0.63 mg kg−1, s.c. Similarly, pretreatment with 15 yielded an ED50 of 0.1 mg kg−1, s.c. The in vivo efficacy of 15 as compared to 13 translated into a lower efficacious plasma concentration (1 nM or approximately 1x the receptor affinity for 15versus 22 nM or 11x the receptor affinity for 13). The higher efficacious plasma concentration for compound 13 was suggestive of an efflux liability as the in vitro MDR evaluation predicted. The lower efficacious plasma concentration for 15 over 13 represented a significant advantage with respect to safety margins. For example, while the HERG channel IC20 for 15 was determined as 104 nM, the ratio of HERG IC20 over plasma efficacious concentration was >200. Alternatively, the HERG IC20 for 13 was 320 nM, yet the ratio of HERG IC20 over plasma efficacious concentration was just 15. On the basis of the in vitro and in vivo evaluation, compound 15 was subsequently profiled in rodent pharmacokinetic studies and in disease relevant models for alcohol intake reduction.
In rats, compound 15 was identified as a high clearance compound (Clb = 120 ml min−1 kg−1) with a substantial volume of distribution (21.7 l) and an 11 h half life. Importantly, it was demonstrated that the compound was metabolized predominantly by CYP enzymes and that the in vivo clearance was effectively predicted by the in vitro clearance values.22 In addition, disposition properties for compound 15 were investigated. Thus, it was demonstrated that 15 readily accessed the central compartment in rats (brain/plasma = 2, at 1 mg kg−1s.c. dosing, 0.75 h time-point). Compound 15 achieved in CSF a concentration of 11.9 nM (1 mg kg−1s.c. dosing, 0.75 h time-point) which equaled the free plasma concentration, thus confirming the in vitro MDR prediction that 15 posed no risk for P-gp efflux (338 nM total plasma concentration, fu of 0.034, 1 mg kg−1s.c. dosing, 0.75 h time-point).
Subsequently, compound 15 was evaluated in a rodent model of measuring reduction in ethanol intake. There are numerous animal models for alcoholism. However, only the selectively bred, alcohol-preferring rats (P-rats) exhibit spontaneous preference for alcohol.23,24 We employed this genetic model of alcoholism as our preclinical model for assessing potential for clinical efficacy. P-rats selectively bred for alcohol preference typically consume in excess of 5 g kg−1 of EtOH per day. Both female and male P rats exhibit this behavior and the intake of female rats is stable across the estrous cycle. In this study, female animals were given free access to food, water and 10% (v/v) alcohol, for 21 days. This was followed by restricted access to alcohol for two hours/day, starting at lights out. Drug was introduced vias.c. administration 30 min prior to the start of this limited access period and the amount alcohol consumed in the drug condition was recorded and compared to baseline intake. Compound 15 produced a dose dependent decrease in alcohol intake (Fig. 3). A dose of 5.6 mg kg−1 produced a 42% reduction in alcohol intake where a dose of 1 mg kg−1 produced a 23% reduction in alcohol intake. This is comparable to the 21% reduction observed at a 3 mg kg−1 dose with the clinically efficacious naltrexone. Thus, compound 15 (CP-866,087) and clinically validated naltrexone both demonstrated efficacy in a preclinical paradigm measuring reduction in alcohol intake.
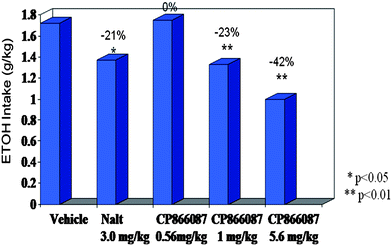 | ||
| Fig. 3 In vivo efficacy of compound 15 in alcohol preferring rats (P-rats). | ||
Conclusions
Compound 15 (CP-866,087) was identified as a potent and selective mu opiate antagonist. The compound was designed with a uniform strategy of targeted lipophilicity reduction. Specifically, it was demonstrated that beta or gamma hydroxylation in a series of aliphatic amines delivered compounds with improved pharmacokinetic parameters including interaction with CYP-2D6. The impact of compound basicity on P-gp efflux was also investigated. Compound 15 demonstrated excellent activity in preclinical models measuring reduction of ethanol intake. On the basis of the preclinical efficacy and its pharmacokinetic performance the compound was selected for clinical evaluation. Results from the clinical performance of this agent will be reported in due course.Notes and references
- (a) D. J. Drobes, R. F. Anton, S. E. Thomas and K. Voronin Alcohol, Alcohol.: Clin. Exp. Res., 2004, 28, 1362 Search PubMed; (b) S. S. O'Malley, Alcohol Alcohol. Suppl., 1996, 31, 77 Search PubMed.
- (a) M. S. Cowen and A. J. Lawrence, Progress Neuro-Psychopharmacol. Biol. Psychiatry, 1999, 23(7), 1171–1212 Search PubMed; (b) R. A. Wise, Pharmacol. Ther., 1987, 35, 227 Search PubMed; (c) T. S. Shippenberg and H. L. Altshuler, Alcohol (Fayetteville, N.Y.), 1985, 2(2), 197 Search PubMed.
- L. G. Latimer, P. Duffy, Kalivas and W. Peter, J. Pharm. Exp. Ther, 1987, 241, 328 Search PubMed.
- A. W. Brunijnzeel, Brain Res. Rev., 2009, 62, 127 Search PubMed.
- (a) D. M. Zimmerman, J. D. Leander, B. E. Cantrell, J. K. Reel, J. Snoddy, L. G. Mendelsohn, B. G. Johnson and C. H. Mitch, J. Med. Chem., 1993, 36, 2833 Search PubMed; (b) J. B. Thomas, S. W. Mascarella, R. B. Rothman, J. S. Partilla, H. Xu, K. B. McCullough, C. M. Dersch, B. E. Cantrell, D. M. Zimmerman and F. I. Carroll, J. Med. Chem., 1998, 41, 1980 Search PubMed.
- B. J. Banks, D. J. Critcher, A. E. Fenwick, D. M. Gethin, S. P. Gibson, U.S. Pat. Appl. Publ. 2003, US 20030207876.
- W. Nagai and Y. Hirata, J. Org. Chem., 1989, 54, 635 Search PubMed.
- D. M. Tschaen, R. Desmond, A. O. King, M. C. Fortin, B. Pipik, S. King and T. R. Verhoeven, Synth. Commun., 1994, 24, 887 CAS.
- L. McMaster and F. B. Langreck, J. Am. Chem. Soc., 1917, 39, 103 Search PubMed.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 10251 CrossRef CAS.
- S. Liras, S. F. McHardy, M. P. Allen, B. E. Segelstein, S. D. Heck, D. K. Bryce, A. W. Schmidt, M. Vanase-Frawley, E. Callegari and S. McLean, Bioorg. Med. Chem. Lett., 2010, 20, 503 CrossRef CAS.
- All compounds gave satisfactory 1HNMR and MS spectral data. (12) 1HNMR (CD3OD) δ 7.34 (t, J = 8.0 Hz, 1H), 7.21–7.27 (m, 3H), 7.10–7.20 (m, 4H), 4.18 (td, J = 6.2, 4.3 Hz, 2H), 3.43 (d, J = 7.0 Hz, 2H), 3.22 (dd, J = 14.5, 7.1 Hz, 2H), 3.10 (d, J = 13.7 Hz, 2H), 2.97 (s, 3H), 2.75–2.92 (m, 3H), 2.36–2.44 (m, 2H), 1.85 (q, J = 7.2 Hz, 2H), 0.92 (t, J = 7.2 Hz, 3H); MS (M + 1) 411.3. (13): 400 MHZ1HNMR (CDCl3) δ 7.16–7.32 (m, 2H), 6.98–7.09 (m, 3H), 2.94–2.97 (m, 4H), 2.81–2.84 (m, 2H), 2.52 (s, 3H), 1.21–1.94 (m, 16H), 0.76 (t, J = 3.4 Hz, 3H); MS (M + 1) 421.2. (14) 400 MHZ1HNMR (CD3OD) δ 7.76 (s, 1H), 7.63–7.65 (m, 1H), 7.41–7.44 (m, 1H), 7.31–7.35 (m, 1H), 7.12–7.14 (m, 2H), 7.01–7.09 (m, 2H), 3.18–3.28 (m, 2H), 3.05–3.10 (m, 2H), 2.98–3.01 (M, 2H), 2.86–2.89 (m, 2H), 2.73 (s, 2H), 1.94–1.99 (m, 2H), 1.81–1.84 (m, 2H), 0.79 (t, J = 7.4 Hz, 3H); MS (M + 1) 377.2. (15) 400 MHZ1HNMR (CDCl3) δ 7.10–7.25 (m, 7H), 6.99–7.05 (m, 1H), 3.19–3.22 (m, 2H), 3.06–3.09 (m, 2H), 2.96–2.99 (m, 7H), 2.81 (s, 2H), 1.83–1.90 (m, 4H), 0.83 (t, J = 7.4 Hz, 3H); MS (M + 1) 427.1. For full experimental procedures please see: S. F. McHardy, S. Liras and S. D. Heck, WO 2003035622, 2003.
- A. S. Kalgutkar, S. Zhou, O. A. Fahmi and T. J. Taylor, Drug Metab. Dispos., 2003, 31, 596 Search PubMed.
- This single point cocktail drug-drug interaction assay determines if a test compound inhibits the 2D6 metabolism of a known substrate (dextromethorophan). The test compound is incubated (3 μM) for 8 min in microsomes and the data are reported as % inhibition of probe substrate metabolism.
- K. M. Giacomini, S.-M. Huang, D. J. Tweedie, L. Z. Benet, K. L. R. Brouwer, X. Chu, A. Dahlin, R. Evers, V. Fischer, K. M. Hillgren, K. A. Hoffmaster, T. Ishikawa, D. Keppler, R. Kim, C. A. Lee, M. Niemi, J. W. Polli, Y. Sugiyama, P. W. Swaan, J. A. Ware, S. H. Wright, S. Wah Yee, M. J. Zamek-Gliszczynski and L. Zhang, Nat. Rev. Drug Discovery, 2010, 9, 215 CrossRef CAS.
- (a) G. J. Diaz, K. Daniell, S. T Leitza, R. L. Martin, Z. Su, J. S McDermott, B. F. Cox and G. A. Gintant, J. Pharmacol. Toxicol. Methods, 2004, 50, 187 Search PubMed; (b) K. Finlayson, L. Turnbull, C. T. January, J. Sharkey and J. S. Kelly, Eur. J. Pharmacol., 2001, 430, 147 Search PubMed.
- S. Liras, S. F. McHardy, M. P. Allen, B. E. Segelstein, S. D. Heck, D. K. Bryce, A. W. Schmidt, R. O'Connor, M. Vanase-Frawley, E. Callegari and S. McLean, Med. Chem. Commun., 2011, 2, 413 RSC.
- Jason D. Hughes, Julian Blagg, David A. Price, Simon Bailey, Gary A. DeCrescenzo, Rajesh V. Devraj, Edmund Ellsworth, Yvette M. Fobian, Michael E. Gibbs, Richard W. Gilles, Nigel Greene, Enoch Huang, Teresa Krieger-Burke, Jens Loesel, Travis Wager, Larry Whiteley and Yao. Zhang, Bioorg. Med. Chem. Lett., 2008, 18, 4872 CrossRef CAS.
- (a) J.-D. Marechal, C. A. Kemp, G. C. K. Roberts, M. J. I. Paine, C. R Wolf and M. J Sutcliffe, Br. J. Pharmacol., 2009, 153, S82 Search PubMed; (b) R. P Sheridan, K. R. Korzekwa, R. A. Torres and M. J. Walker, J. Med. Chem., 2007, 50, 3173 CrossRef CAS; (c) S. Sciabola, I. Morao and M. J. de Groot, J. Chem. Inf. Model., 2007, 47, 76 Search PubMed; (d) C. A Kemp, J. U. Flanagan, A. J. van Eldik, J.-D. Marechal, C. R. Wolf, G. C. K. Roberts, M. J. I. Paine and M. J. Sutcliffe, J. Med. Chem., 2004, 47, 5340 Search PubMed; (e) D. F. V. Lewis, P. J. Eddershaw, P. S. Goldfarb and M. H. Tarbit, Xenobiotica, 1997, 27, 319 Search PubMed.
- C. R Johnson and B. D. Tait, J. Org. Chem., 1987, 52, 281 CrossRef CAS.
- Conformational search was performed with MacroModel module in Maestro software package (V. 9.1) (Schrodinger Inc, NYC, NY). The lowest energy conformations for both neutral and protonated forms were subjected to further calculation. The COSMO charges of the lowest energy conformations were calculated with the CALCULATE program from COSMOLogic using the BP-TZVP-COSMO method (b–p function and def-TZVP basis set). The pKa of the simplified analogs of 13 and 15 were calculated based on the COSMO charge files in COSMOThermX.
- N. A. Hosea, W. T. Collard, S. Cole, T. S. Maurer, R. X. Fang, H. Jones, S. M Kakar, Y. Nakai, B. J. Smith, R. Webster and K. Beaumont, J. Clin. Pharmacol., 2009, 49, 513 Search PubMed.
- T. K Li, L. Lumeng, W. J. McBride and J. M. Murphy, Alcohol Alcohol Suppl, 1987, 1, 91 Search PubMed.
- Animals were handled and cared for in accordance with the guide for the care and use of laboratory animals (National Research Council 1996) and all procedures were performed with the approval of the Pfizer Animal Care committee.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00164g |
| ‡ Current Address: Synthesis and Process Chemistry, Southwest Research Institute, 6220 Culebra Rd, San Antonio, Texas, 78238, USA. |
| This journal is © The Royal Society of Chemistry 2011 |