Optimized glucuronidation of dual pharmacology β-2 agonists/M3 antagonists for the treatment of COPD†
Laura
Hilton
a,
Rachel
Osborne
a,
Amy S.
Kenyon
a,
Helen
Baldock
a,
Mark E.
Bunnage
a,
Jane
Burrows
b,
Nick
Clarke
c,
Michele
Coghlan
c,
David
Entwistle
b,
David
Fairman
d,
Neil
Feeder
b,
Kim
James
a,
Rhys M.
Jones
d,
Nadia
Laouar
a,
Graham
Lunn
a,
Stuart
Marshall
c,
Sandra D.
Newman
a,
Sheena
Patel
c,
Matthew D.
Selby
a,
Fiona
Spence
e,
Emilio F.
Stuart
c,
Susan
Summerhill
c,
Michael A.
Trevethick
c,
Karen N.
Wright
c,
Michael
Yeadon
c,
David A.
Price
a and
Lyn H.
Jones
*a
aSandwich Chemistry, World Wide Medicinal Chemistry, Pfizer, Ramsgate Road, Sandwich, CT13 9NJ, UK
bPharmaceutical Sciences, Pfizer, Ramsgate Road, Sandwich, CT13 9NJ, UK
cAllergy and Respiratory Biology, Pfizer, Ramsgate Road, Sandwich, CT13 9NJ, UK
dPharmacokinetics, Dynamics and Metabolism, Pfizer, Ramsgate Road, Sandwich, CT13 9NJ, UK
eDrug Safety, Pfizer, Ramsgate Road, Sandwich, CT13 9NJ, UK
First published on 11th July 2011
Abstract
‘Inhalation by design’ concepts were developed to create novel dual pharmacology β-2 agonists-M3 antagonists, for the treatment of chronic obstructive pulmonary disorder. A key feature of this work is the combination of balanced potency and pharmacological duration with optimised glucuronidation through the incorporation of metabolically vulnerable phenols.
Chronic obstructive pulmonary disease (COPD) is caused by emphysema and chronic bronchitis that damages the airways of the lungs leading to significantly reduced air flow and is predicted to be the third largest cause of death by 2030.1 Treatments for this debilitating disease include the use of bronchodilators, the two major classes being inhaled β-2 adrenergic agonists and muscarinic M3 antagonists. The combination of long acting β-2 agonists (LABAs) and long acting muscarinic antagonists (LAMAs) have enabled once-daily inhalation dosing regimens to be optimized.2
Further improvements in treatment can be realized through triple therapies that combine a LABA, LAMA and an inhaled corticosteroid.3 However, the complexity of combining three different drugs that operate via three distinct mechanisms into a single device for inhalation dosing, such that patient compliance is high, is considerable. To facilitate the triple therapy concept, we, and others,4 have pursued a strategy to incorporate muscarinic antagonism and β-2 agonism into a single molecule (MABA), such that combination with an ICS could be achieved in a dry powder inhaler.
We have recently described our creation of tolterodine-containing MABAs, where balanced potency and pharmacological duration was combined with high metabolic clearance, low oral bioavailability and desirable material properties, culminating in compound 1 (Fig. 1).4c
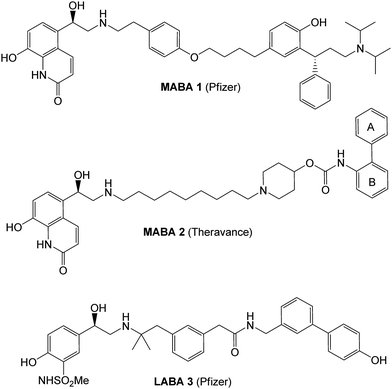 | ||
| Fig. 1 Inhaled bronchodilators: MABAs 1 and 2, and LABA 3. | ||
As an alternative strategy, we also pursued MABAs containing biaryl carbamoyl piperidine antimuscarinics linked to the sulfonamide β-2 head group, in a manner conceptually similar to that previously reported by Hughes et al. that furnished molecules such as 2 (Fig. 1).4d,e We were keen to explore the potential of incorporating additional modes of metabolism into our design to further maximize clearance and mitigate any potential issues for drug-drug interactions (DDIs). The phase II metabolic pathway of glucuronidation seemed ideally suited to our task due to its high turnover capacity and the rapid elimination of the resulting glycosylated metabolites. Although carboxylic acids are well precedented substrates for uridine-5′-diphosphoglucuronosyl transferase (UGT), the potential to yield reactive acyl-glucuronide conjugates raised concerns regarding potential safety issues,5 and therefore our preference was to pursue the phenol moiety as an alternative metabolically vulnerable motif. Indeed, this strategy was used successfully in the design of LABA 3 (Fig. 1) and related analogues in this series by Glossop and co-workers at Pfizer, Sandwich.6 We were also keen to retain the sulfonamide β-2 head group as this appeared to impart crystallinity to the resultant LABA molecules, and the presence of significant quantities of late stage intermediates within Pfizer greatly facilitated the creation of novel MABAs in this class.
As a standard and benchmark for this work, we measured the glucuronidation rate for the Theravance MABA 2. In human liver microsomes incubated in the absence of NADPH and presence of the microsomal activator Brij58 and UDPGA (uridine-5′-diphosphoglucuronic acid),72 exhibits a small intrinsic clearance of 9 μl/min/mg protein by the glucuronidation pathway, presumably due to turnover of the head group phenol (Table 1).8
| Compound | β-2 EC50 (nM)b | M3 Ki (nM)c | Glucuronidation (μl/min/mg)d | HLM e (μl/min/mg) | cLogPf |
|---|---|---|---|---|---|
| a nd = not determined. All molecules possess selectivity over β1 (EC50 > 10 μM). b potency of compounds at the β-2 adrenoceptor, that measures their ability to stimulate intracellular cAMP production in CHO-hβ-2 cells. c potency of compounds at the human cloned M3 receptor using [3H] N-methyl scopolamine. d glucuronidation rate assessed in HLM (-NADPH +Brij, +UDPGA). e clearance values in human liver microsomes. f calculated LogP. | |||||

|
13 | 0.82 | 9.0 | 51 | 5.6 |

|
74 | 0.39 | <2 | 40 | 5.3 |

|
26 | 2.2 | 4 | 20 | 4.6 |

|
129 | 3.7 | <3.5 | 26 | 4.6 |
|
90 | 18 | <2 | 17 | 4.3 |
|
nd | 26 | <2 | 8.8 | 4.6 |

|
nd | 10 | <2 | nd | 4.8 |

|
nd | >73 | 3.7 | 33 | 4.5 |

|
102 | 22 | <3.5 | 20 | 4.8 |

|
65 | 76 | 4 | 28 | 5.1 |

|
26 | 5.0 | 30 | 16 | 4.9 |
|
22 | 8.3 | 90 | 13 | 5.1 |

|
3.7 | 0.73 | 24 | 23 | 5.3 |
|
nd | 4.8 | 19 | 22 | 5.6 |

|
14 | 2.0 | 6 | 29 | 5.6 |

|
36 | 38 | 32 | 79 | 5.6 |

|
23 | 1.1 | 17 | 25 | 5.7 |
|
57 | 4.4 | 6 | 15 | 5.7 |
|
20 | 1.1 | 24 | 17 | 4.8 |
|
21 | 2.0 | 22 | 34 | 5.8 |

|
195 | 4.4 | 16 | 45 | 6.4 |

|
24 | 9.2 | 51 | 39 | 5.5 |
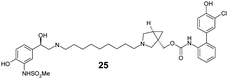
|
17 | 4.3 | 28 | 49 | 5.6 |

|
42 | 1.3 | 13 | 7 | 5.1 |
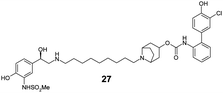
|
23 | 8.9 | 7.8 | 8.0 | 5.6 |

|
nd | 36 | 48 | 74 | 4.3 |
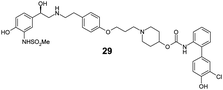
|
13 | 3.1 | 29 | 33 | 4.6 |
The sulfonamide analogue 4 was prepared (using similar chemistry to that previously described)4d,e and found to retain dual pharmacology (although somewhat less potent versus β-2 relative to 2), but was resilient to glucuronidation in the above assay. As a result, phenols were introduced into the antimuscarinic motif, whilst still trying to retain (and balance) the requisite dual pharmacology. Our aim here was also to balance the modes of metabolism (i.e. whereby the HLM and UGT turnovers would ideally be of comparable values).
These analogues were efficiently prepared using the divergent synthetic strategy exemplified in Scheme 1 for derivative 15.4f Briefly, carbamate 31 was prepared through the addition of Boc-protected hydroxyl piperidine 30 and 2-bromophenylisocyanate (both commercially available). Acid-mediated deprotection of the Boc group, to provide 32, enabled installation of the linker moiety using high dilution alkylation conditions. Amine deprotection gave 33 and this allowed conjugation of the β-2 head group sulfonamide 34 and the intermediate 35 was prepared on scale to furnish a number of derivatives modified in the A-ring through Suzuki coupling of aryl boronic acids (36). Hydrogenolysis of the benzyl protecting group and silyl deprotection yielded the final compound 15. This divergent route facilitated the preparation of numerous analogs in this MABA series (see Supplementary Information).
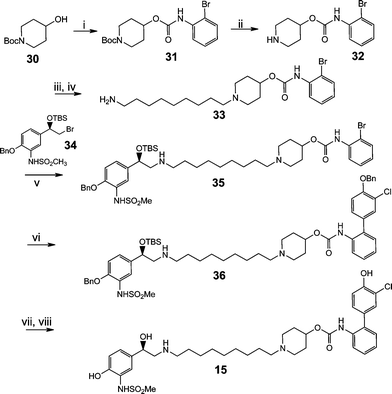 | ||
| Scheme 1 Divergent synthetic strategy to prepare MABA analogues such as 15. i) 2-BrPhNCO, CH2Cl2, NEt3, 68%; ii) 4M HCl aq., 16 h, 84%; iii) (Boc)2N(CH2)8CH2Br, NaHCO3, CH3CN, 90 °C, 16 h, 71%; iv) 4M HCl aq., 16 h, 95%; v) CH3CN, NaHCO3, 90 °C, 16 h, 61%; vi) Pd(OAc)2, P(o-tolyl)3, NEt3, 120 °C, 3 h, 92%; vii) tButylmethyl ether, 10% Pd/C, 6 h, 15 psi H2, 88%; viii) Et3N(HF)3, THF, 3 h, 96%. | ||
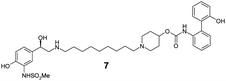
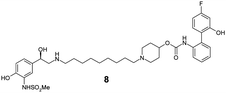
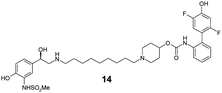
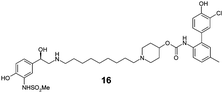
The incorporation of phenols into the A-ring (see MABA 2, Fig. 1) to provide 5, 6 and 7 did not introduce significant glucuronidative metabolism, but did point to the para-substitution being preferred for β-2 and M3 potency (compound 5, EC50 26 nM and Ki 2.2 nM respectively). Next, we attempted to enhance/switch on glucuronidation of the MABA through the introduction of halogen atoms to increase the acidity of the phenol (and similarly through the incorporation of a nitrile group in compound 10).9 Although this strategy was unsuccessful for the ortho- and meta-phenols (8–12), a breakthrough for the project was realized when this approach was applied to the para-substituted phenol as this provided a significant increase in glucuronidation rates, whilst maintaining the desirable dual pharmacology (13, 14, 15). Indeed, the substitution pattern of the chloro-phenol 15 seemed an optimal balance of metabolic vulnerability and dual pharmacology and this group was retained for further work. Additional structure–activity/metabolism relationships were generated through modification of the B-ring, particularly where the effect of increased lipophilicity may improve metabolic turnover, but this met with limited success (16–20).
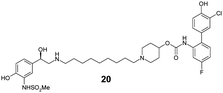
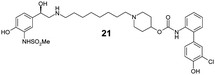
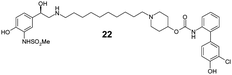
Finally, we investigated the effect of different linkers on the medicinal chemistry profile of these derivatives. Tether length changes had little effect on glucuronidation (21–23, presumably due to the β-2 motif extending well-beyond the binding/reaction site), but reduced the β-2 and M3 potency relative to compound 15. The observation that the C9-linker is optimal for dual pharmacology is in line with observations by Theravance and may reflect a multivalent effect.4d,10 Replacement of the piperidine moiety with pyrrolidine (24) appeared to increase glucuronidation (though compromised M3 potency), whilst the 3-aza-bicyclo[3.1.0]hexane linkers in 25 and 26 seemed to have little advantage over piperidine, particularly as it increased synthetic complexity of the resulting MABAs. Similarly, the 8-azabicyclo[3.2.1]octane linker present in 27 showed no improvement over 15. Incorporation of phenol ether linkers, as present in 1, was found to increase glucuronidation (28, 29), but they have a detrimental effect on potency relative to compound 15. It is interesting to note that subtle changes in the linker structure are able to modify the rates of glucuronidation, suggesting these regions do contact the UGT enzymes, but further work is required to fully delineate these effects.
Metabolite identification in human hepatocytes using LC/MS/MS confirmed the presence of the phenol glucuronide (and sulfate) of 15, thus providing further evidence for phase II elimination. Since compound 15 was obviously the outstanding MABA from this series, it was progressed to further in vitro profiling and into our COPD models of bronchoconstriction.
The kinetics of pharmacological duration were assessed for β-2 through a simple receptor wash-off assay as previously described,11 and for the M3 component using a dilution-offset methodology whereby the offset is inferred from the on rate of co-administered 3H-NMS (N-methyl scopolamine) and is expressed as the time taken to reach 50% of total 3H-NMS for solvent treated membranes.12 Pleasingly, values were indicative of long residency at both receptors (Table 2).13
| β-2 wash off-fold shiftb | M3 offset (min)c | GPT EFS EC50 (nM)d | GPT EFS duration T50% (hr)d | GPT EFS +propranolol EC50 (nM)e | GPT EFS +propranolol duration (hr)e | GPT histamine EC50 (nM)f |
|---|---|---|---|---|---|---|
| a All data are geometric means with 95% CI, n = 4. b fold-difference in β-2 potency following receptor wash. The long- acting β-2 agonist salmeterol has a shift of 2.2-fold in this assay. c offset kinetics from human cloned M3 receptor using [3H] N-methyl scopolamine (dilution method). The short acting inhaled antimuscarinic ipratropium has an offset of 10 min in this assay. d GPT = Guinea pig trachea. EFS = Electric field stimulation. Potency and duration for β-2 and M3 components of bronchodilation. Submaximal concentration used for the duration is that which gives 70% inhibition of the EFS response. e Potency and duration for M3 component of bronchodilation. f Potency for β-2 component of bronchodilation. | ||||||
| 1.5 | 117 | 7.3 | >11 | 12 | >11 | 5.8 |
We then assessed the potency of 15 in the guinea-pig trachea (GPT) model using electric field stimulation to release endogenous acetylcholine.14 The drive for bronchoconstriction in this model operates through both β-2 and M3 mechanisms and therefore values here reflect combined bronchodilatory effects. A ‘duration of action’ was defined as the time taken for the muscle tone at a submaximal concentration of the compound to recover by 50% of the inhibition induced. As expected, 15 is a potent bronchodilator in this model with respectable duration of action (Table 2). To delineate the M3 drive, the β-2 antagonist propranolol was added to the EFS model to block the β-2 effect. 15 retained both potency and duration of action as predicted from the in vitro residency experiments. To confirm the β-2 pharmacological component, the GPT assay was modified to use histamine as the bronchoconstricting agent.1515 was a potent bronchodilator in this model as expected.16
15 effected a dose-related inhibition of methacholine-induced bronchoconstriction in dogs16 when dosed intratracheally, with an ID50 of 50 μg, 14–16 h duration of action consistent with bid dosing and a therapeutic index over cardiovascular side effects (heart rate and contractility) of 20 (salmeterol TI = 3–10 in this model). The CV effects are likely driven by the β-2 primary pharmacology since 15 was found to be inactive versus β-1 (EC50 > 10 μM). 15 was also confirmed as possessing single and dual pharmacology in conscious and anaesthetized guinea pigs with a potency and duration of action similar to salmeterol.17 Results in dog and guinea pig with a continuous infusion of propranolol confirmed the M3 component of bronchodilation in vivo, while the β-2 component was confirmed when the NK2 agonist BANA was used as bronchoconstrictor.
Additionally, 15 was clean in in vitro genetic toxicity testing (AMES and micronucleus) and was also found to have a clean irritancy profile in a rabbit cough model developed at Sandwich (assessment of Aδ-fibre firing in the vagus nerve), giving confidence that it would be a low cough risk in humans.18
Rat and guinea pig pharmacokinetics experiments on 15,19 when combined with the poor membrane permeability, metabolic instability, and optimized glucuronidation, predict negligible human oral bioavailability (<5%) thus minimizing potential risks from systemic exposure.
As the product profile of a MABA requires formulation into a dry powder inhaler, either as a standalone agent, or as a combination therapy with an ICS, it is imperative to have the desired material properties. Following an extensive sitting-drop salt screen,2015 was crystallized as the naphthalene 1,5-disulfonate hydrate salt (melting point 200 °C), thus enabling further formulation development of this compound. Jet milled material (3–5 μm diameter by scanning electron microscopy) was blended with lactose monohydrate (1/100 weight ratio), the most common carrier excipient used in commercial dry powder inhalers. Accelerated stability testing of this material (70 °C, 75% relative humidity, 7 days) showed that no single impurity increased to beyond the 0.3% level.
Conclusions
Several novel MABAs were created using the principles of ‘inhalation by design’. Balanced potency and pharmacological duration was combined with optimal phase II metabolismvia glucuronidation. The general learnings provided by the structure-metabolism relationships generated in this work should prove useful for both inhaled and oral drug discovery programmes, depending on whether glucuronidation is a feature to design in, or to avoid. However, a detailed rationalisation of these relationships is currently hindered by complications arising from UGT subtype heterogeneity, multiple binding/reaction sites and differences in glucuronidation kinetics.Combined with the high metabolic clearance, low synthetic complexity (by virtue of the simple C9-methylene linker), low oral bioavailability, desirable material properties and impressive TI over haemodynamic effects, these characteristics led to the nomination of compound 15 as a clinical candidate (PF-4348235). This work should significantly advance triple therapy paradigms for COPD through combination with inhaled corticosteroids. Further elaboration and development of this series, including a detailed description of the pharmacology of MABA 15 and its optimized synthetic route to provide kilogram quantities of material, will appear elsewhere.
Acknowledgements
We thank Paul Glossop and Alan Stobie for useful discussions and Louise Sladen for measuring cough potential.Notes and references
- http://www.who.int/respiratory/copd/en/ .
- M. Cazzola and M. Molimard, Pulm. Pharmacol. Ther., 2010, 23, 257 Search PubMed.
- T. Welte, Int. J. Clin. Pract., 2009, 63, 1136 Search PubMed.
- (a) N. Ray and L. Alcaraz, Expert Opin. Ther. Pat., 2009, 19, 1 Search PubMed; (b) M. Cazzola and M. Matera, Br. J. Pharmacol., 2009, 155, 291 CrossRef; (c) L. H. Jones, H. Baldock, M. Bunnage, J. Burrows, N. Clarke, M. Coghlan, D. Entwistle, D. Fairman, N. Feeder, C. Fulton, L. Hilton, K. James, R. Jones, A. Kenyon, S. Marshall, S. Newman, R. Osborne, S. Patel, M. Selby, E. Stuart, M. Trevethick, K. Wright and D. Price, Bioorg. Med. Chem. Lett., 2011, 21, 2759 CrossRef CAS; (d) A. Hughes, K. Chin, S. Dunham, J. Jasper, K. King, T. Lee, M. Mammen, J. Martin and T. Steinfeld, Bioorg. Med. Chem. Lett., 2011, 21, 1354 CrossRef CAS; (e) M. Mammen, S. Dunham, A. Hughes, W. Tae, C. Husfeld and E. Stangeland, US patent 20040167167; (f) L. H. Jones, G. Lunn and D. Price, WO patent 2008041095.
- S. Regan, J. Maggs, T. Hammond, C. Lambert, D. Williams and B. Park, Biopharm. Drug Dispos., 2010, 31, 367 CrossRef CAS.
- P. Glossop, C. Lane, D. Price, M. Bunnage, R. Lewthwaite, K. James, A. Brown, M. Yeadon, C. Perros-Huguet, M. Trevethick, N. Clarke, R. Webster, R. Jones, J. Burrows, N. Feeder, S. Taylor and F. Spence, J. Med. Chem., 2010, 53, 6640 CrossRef CAS.
- O. Trubetskoy, M. Finel and V. Trubetskoy, J. Pharm. Pharmacol., 2008, 60, 1061 CrossRef CAS.
- T. Shimizu, H. Mori, E. Tabusa, G. Miyamoto, Y. Yasuda and K. Nakagawa, Xenobiotica, 1978, 8, 349 CrossRef CAS.
- B. Ethell, S. Ekins, J. Wang and B. Burchell, Drug Metab. Dispos., 2002, 30, 734 CrossRef CAS.
- T. Steinfeld, A. Hughes, U. Klein, J. Smith and M. Mammen, Mol. Pharmacol., 2010, 79, 389.
- S. Summerhill, T. Stroud, R. Nagendra, C. Perros-Huguet and M. Trevethick, J. Pharmacol. Toxicol. Methods, 2008, 58, 189 CrossRef CAS.
- J. Watson, M. Strawbridge, R. Brown, K. Campany, M. Coghlan and M. Trevethick, Fund. Clin. Pharm, 2008, 22(Suppl. 2), 69 Search PubMed.
- S. Patel, S. Marshall, S. Summerhill, M. Coghlan, M. Strawbridge, M. Stanley, E. Stuart, N. Clarke, M. Trevethick, C. Perros-Huguet and M. Yeadon, Am. J. Respir. Crit. Care Med, 2011, 183, A1607 (poster).
- A. Brown, M. Bunnage, P. Glossop, K. James, R. Jones, C. Lane, R. Lewthwaite, S. Mantell, C. Perros-Huguet, D. Price, M. Trevethick and R. Webster, Bioorg. Med. Chem. Lett., 2007, 17, 4012 CrossRef CAS.
- F. Roux, B. Grandordy and J. Douglas, Am. J. Respir. Crit. Care Med, 1996, 153, 1489 Search PubMed.
- S. Patel, S. Marshall, S. Summerhill, M. Strawbridge, M. Stanley, C. Fiddler, E. Stuart, N. Clarke, M. Trevethick, C. Perros-Huguet and M. Yeadon, Am. J. Respir. Crit. Care Med, 2011, 183, A1608 (poster).
- (a) K. Hulland, J. Philip, I. Delescluse, N. Clarke, C. Perros-Huguet and M. Yeadon, Am. J. Respir. Crit. Care Med, 2011, 183, A1610 (poster); (b) J. Philip, K. Hulland, N. Clarke, C. Perros-Huguet and M. Yeadon, Am. J. Respir. Crit. Care Med, 2011, 183, A1609 (poster).
- L. Sladen, N. Clarke, C. Perros-Huguet and M. Yeadon, Am. J. Respir. Crit. Care Med, 2011, 183, A3099 (poster).
- Rat iv pharmacokinetics (dose 2 mg/Kg): AUC 125 ng hr/mL; clearance 164 mL/min/Kg. Rat oral (dose 0.2 mg/Kg): AUC 256 ng hr/mL; clearance 14 mL/min/Kg; T1/2 1.6 h. Guinea pig iv (dose 0.2 mg/Kg): AUC 920 ng hr/mL; clearance 3.6 mL/min/Kg; T1/2 0.8 h.
- D. Clark, Am. Pharm. Rev., 2004, 7, 76 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed preparation of 15 and characterization data for all MABAs, 4–29. See DOI: 10.1039/c1md00140j |
| This journal is © The Royal Society of Chemistry 2011 |