Development of substituted 7-phenyl-4-aminobenzothieno[3,2-d] pyrimidines as potent LIMK1 inhibitors†
Brad E.
Sleebs
abc,
Danny
Ganame
abc,
Alla
Levit
abc,
Ian P.
Street
abc,
Alison
Gregg
d,
Hendrik
Falk
abc and
Jonathan B.
Baell
*abc
aThe Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria, Australia 3052. E-mail: jbaell@wehi.edu.au; Tel: +613 9345 2108
bCancer Therapeutics-CRC P/L, 4 Research Ave, La Trobe R&D Park, Bundoora, Victoria, Australia 3086
cDepartment of Medical Biology, The University of Melbourne, Parkville, Victoria 3010, Australia
dCentre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), 381 Royal Parade, Parkville, 3052, Australia
First published on 12th August 2011
Abstract
7-Phenyl-4-aminobenzothieno[3,2-d]pyrimidines were previously reported to exhibit moderate LIMK1 inhibition. Further exploration of SAR around the 7-phenyl moiety has led to the development of a lead series with an increased potency for LIMK1. Evaluation of physicochemical and ADME properties, and off-target kinase screens has seen this novel series emerge as a promising platform for a set of tool compounds for evaluating LIMK as a therapeutic target.
Introduction
The LIMK family of proteins are serine protein kinases comprised of two proteins, LIMK1 and LIMK2 (LIMK stands for LIM-kinase, where LIM is an acronym of the three gene products Lin-11, Isl-1 and Mec-3). They are recognised by two common LIM domains, along with a PDZ motif and a catalytic domain. LIMK 1 and 2 overall share approximately 50% homology, with 70% identity in the catalytic domain.1LIMK proteins are involved in regulation of actin polymerisation and microtubule disassembly. LIMK1 and 2 are regulated themselves by phosphorylation by PAK1, PAK2 and Rho kinases. LIMK proteins, and closely related TESK, are the only known kinases to phosphorylate and inactivate cofilin, resulting in polymerisation of globular actin into filamentous actin. The consequence is the reorganisation of the filamentous actin cytoskeleton, and regulation of cell morphology and motility.2
In tumour cells, the normal mechanisms that control the actin cytoskeleton dynamics undergo changes that allow the cell to become metastatic and invasive. Studies have shown that LIMK1 plays a critical role in metastasis. LIMK1 has been found to be highly expressed in a number of malignant tumour cell lines.1 Further, studies have shown that knockdown models of LIMK1 or overexpression of cofilin leads to a decrease in motility of highly metastatic cell lines.1 These studies have led to the belief that disruption of the dynamic equilibrium between cofilin and its phosphorylated form will result in reduced metastasis and invasiveness. Hence the development of a small molecule inhibitor of LIMK1 will not only provide insights into the precise role of LIMK1, but may provide a novel approach to cancer therapy. Inhibition of LIMK2 is believed not to play a critical role in metastasis, but rather in cell cycle progression.2
Thus far, there have been a limited number of LIMK1 inhibitors described in the literature.3 Bristol Myers Squibb (BMS) disclosed two classes of inhibitors, a pyrazolo series and a 5-thiazolopyrimidine series.4–6 Some members of the pyrazolo series were shown to be potently cytotoxic but it was determined that this was due to off-target (anti-microtubule) activity and that LIMK inhibition itself did not result in inhibition of cellular proliferation.4 As BMS were not interested in an anti-metastatic program, the series development was halted. More recently Lexicon pharmaceuticals developed a pyrrolopyrimidine series as potent inhibitors of LIMK1 and 2 for treatment of ocular hypertension and glaucoma.7–9 Results from this study are ongoing.
Our interest lies in the development of LIMK1 inhibitors as anti-metastatic agents, although we also had evidence that inhibition in vivo could diminish primary tumour growth. We recently reported that a high throughput screen (HTS) of a diverse 40000 compound library led to the identification of the 3-aminothieno[2,3-b]pyridine-2-carboxamide 1 with weak LIMK1 inhibitory activity.10 Transformation to aminopyridine 3 gave an appreciable increase in activity and ring closure of other systems such as 2 to 4 did so similarly. We also found that indole-based systems 5 were well tolerated. Finally a survey of phenyl substitution around the core led to the 7-phenyl analogue 6, which exhibited a 20-fold improvement in activity against LIMK1. A similar improvement was also displayed with the 7-phenyl indole derivative 7 (Fig. 1 and Table 1).10 Herein, we report on the further development of 7-pheny-4-aminobenzothieno[3,2-d]pyrimidines as LIMK1 inhibitors, with a focus on making these hydrophobic compounds more druglike by incorporating polar substituents on the 7-phenyl ring.
| Compound | IC50, nM | Compound | IC50 (nM) |
|---|---|---|---|
| a KinaseGlo assay format (see supplementary information†). | |||
| 1 | 37,000 | 5 | 4,000 |
| 2 | 33,000 | 6 | 300 |
| 3 | 9,000 | 7 | 260 |
| 4 | 7,000 |
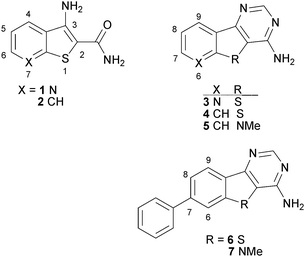 | ||
| Fig. 1 Previous progression summary of LIMK1 inhibitors. | ||
As shown in Scheme 1, the first set of SAR probes were readily made via a Suzuki reaction on the bromo precursor 8, and structures and biological data for 31 of these are given in Table 2.
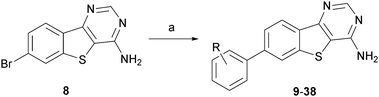 | ||
| Scheme 1 Synthesis and structures of compounds 9–38. Reagents and conditions: a) substituted phenylboronic acid, PdCl2(PPh3)2, K2CO3, dioxane, H2O, 90 °C. | ||
| Compound | Ph Substitutionb | nc | IC50 (nM) |
|---|---|---|---|
| a KinaseGlo assay format (see supplementary information†). b An N′ signifies an endocyclic nitrogen atom. c Number of measurements. | |||
| 6 | none | 11 | 300 |
| 9 | 2-NH2 | 2 | 310 |
| 10 | 2-OH | 2 | 260 |
| 11 | 2-NHSO2Me | 2 | 998 |
| 12 | 2-Cl | 2 | >1000 |
| 13 | 2-OMe | 2 | 220 |
| 14 | 3-NH2 | 1 | 231 |
| 15 | 3-OH | 2 | 230 |
| 16 | 3-Cl | 1 | 199 |
| 17 | 3-OMe | 2 | 149 |
| 18 | 3-CONH2 | 5 | 120 |
| 19 | 3-COOH | 2 | 196 |
| 20 | 3-tetrazole | 4 | 50 |
| 21 | 3-SO2Me | 1 | 361 |
| 22 | 3-CH2OH | 2 | 129 |
| 23 | 3-NHCOH | 11 | 55 |
| 24 | 3-NHAc | 2 | 395 |
| 25 | 3N′ | 1 | 795 |
| 26 | 4-NH2 | 2 | 159 |
| 27 | 4-OH | 2 | 267 |
| 28 | 4-NHSO2Me | 2 | 243 |
| 29 | 4-OMe | 2 | 204 |
| 30 | 4-CONH2 | 2 | 156 |
| 31 | 4-tetrazole | 1 | 127 |
| 32 | 4-SO2Me | 1 | 376 |
| 33 | 4-CH2OH | 1 | 163 |
| 34 | 4-CN | 1 | 828 |
| 35 | 4-Ac | 1 | 172 |
| 36 | 4-NHCOH | 4 | 44 |
| 37 | 4-NHAc | 3 | 181 |
| 38 | 4N′ | 1 | 732 |
From the data presented in Table 2, it can be seen that substitution at the 2-position can be tolerated if an NH2 (9, IC50 310 nM), an OH (10, IC50 260 nM), or an OMe (13, IC50 220 nM), but no real boost in potency is observed. A bulkier polar group as NHSO2Me leads to a loss of activity (11, IC50 998 nM) as does an electron-withdrawing hydrophobic substituent such as Cl (12, IC50 >1000 nM).
Likewise for the 3-position, a number of groups can be tolerated, including NH2 (14, IC50 231 nM), OH (15, IC50 230 nM), but less so SO2Me (21, IC50 361 nM) or NHAc (24, IC50 395 nM) or an aza-carba replacement (25, IC50 795 nM). However, in contrast to the 2-position, a 3-Cl leads to a slight improvement in activity (16, IC50 199 nM), as does a 3-OMe (17, IC50 149 nM), CONH2 (18, IC50 120 nM), COOH (19, IC50 196 nM), and a CH2OH (22, IC50 129 nM). Intriguingly, a marked improvement in activity is observed with a 3-tetrazole (20, IC50 50 nM), or 3-NHCOH (23, IC50 55 nM). Since a 3-Cl is quite different to a formamide group, it is plausible that different binding modes in the ATP binding site are involved (we have determined that this class is ATP-competitive), especially as a meta-substituent can sample a significant amount of topographical space.
In the 4-position, it is also the case that a variety of groups are tolerated, such as OH (27, IC50 267 nM), NHSO2Me (28, IC50 243 nM), OMe (29, IC50 204 nM) and SO2Me (32, IC50 376 nM), while others, such as CN (34, IC50 828 nM) or an endocyclic nitrogen atom (38, IC50 732 nM), are not.
However, there is a definite increase in activity with some substituents, such as NH2 (26, IC50 159 nM), CONH2 (30, IC50 156 nM), tetrazole (31, IC50 127 nM), CH2OH (33, IC50 163 nM), Ac (35, IC50 172 nM), NHAc (37, IC50 181 nM). In particular, the NHCOH 36 displayed an IC50 of 44 nM. Once again, some of this SAR is hard to interpret in association with a single binding mode, and it is plausible that different SAR streams are involved here.
Given the promising activity of benzamide 18 and anilide 23, we made a series of ureas and amides in the 3-position to further sample binding site space. Formation of the ureas was performed by reaction of the 3-amino substituted arene 14 with commercially available isocyanates. When the isocyanate was not readily available a two step process was undertaken. This first involved reacting 14 with phenyl chloroformate forming the activated carbamate. Subsequent reaction with the appropriate amine afforded the substituted urea (Scheme 2). The synthesis of the amides was performed from the 3-carboxylic acid 19, via a HBTU mediated amide coupling (Scheme 3). All compounds were evaluated for LIMK1 inhibition and the results are summarised in Tables 3 and 4.
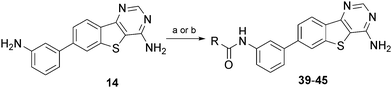 | ||
| Scheme 2 Synthesis and structures of compounds 39–45. Reagents and conditions: a) RNCO, dioxane; b) i) PhOCOCl, DIPEA; ii) amine, dioxane, 80 °C. | ||
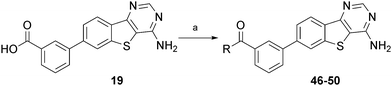 | ||
| Scheme 3 Synthesis and structures of compounds 46–50. Reagents and conditions: a) HBTU, DIEPA, amine, DMF. | ||
In the urea series, some extensions were quite unfavourable, such as for the isobutylurea 42 (IC50 237 nM) and extended piperazinyl urea 43 (IC50 223 nM). However, while we did not observe a further boost in potency relative to 23, several compounds displayed IC50 values of less than 100 nM, such as N-ethylurea 40 (IC50 87 nM), N-isopropylurea 41 (IC50 78 nM), the pyrrolidinyl urea 44 (IC50 95 nM) and the N-methyl piperazinyl urea 45 (IC50 95 nM).
In the amide series, several compounds lost some activity, such as the ethanolamide 46 (IC50 228 nM), the alaninamide 47 (IC50 294 nM) and the morpholinamide 49 (IC50 >1000 nM), but some compounds were quite potent, such as the morpholine glycinamide 48 (IC50 81 nM), and the 3-ethylenepyridinamide 50 (IC50 97 nM). We were particularly interested in compounds that were both potent and potentially more water soluble, such as 45, 48 and 50.
A small subset of 7-phenylindole compounds 52–54 was also synthesised to ascertain whether the SAR pattern mirrored the 7-phenylbenzothiophene series compounds 27, 18 and 36. A Suzuki reaction of the 7-bromoindole 51 produced four analogues and these compounds were tested for LIMK1 inhibition (Scheme 4 and Table 5). From these data it was observed that the SAR between the two series did not crossover. Notably, the indole 7-phenyl-3′-carboxamide 53 has an IC50 of 379 nM, compared with its benzothiophene 18 comparator of 120 nM. Similarly, the indole 7-phenyl-4′-NH-formyl 54 has an IC50 of 498 nM, compared with its benzothiophene 36 comparator of 44 nM. We explain this as follows: superimposition of the pyrimidine ring in the two classes leads to a clearly different disposition of the 7-phenyl ring due to the longer carbon-sulfur bonds. This may not be too influential when the 7-phenyl ring is unsubstituted (and so 6 and 7 are similar in activity), but is likely to be more so for the substituents off the 7-phenyl ring.
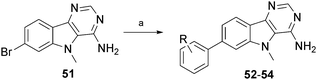 | ||
| Scheme 4 Synthesis and structures of compounds 52–54. Reagents and conditions: a) substituted phenylboronic acid, PdCl2(PPh3)2, K2CO3, dioxane, H2O, 90 °C. | ||
| Compound | Ph Substitution | n | IC50 (nM) |
|---|---|---|---|
| a Transcreener assay format (see supplementary information†). | |||
| 7 | none | 4 | 260 |
| 52 | 4-OH | 2 | 571 |
| 53 | 3-CONH2 | 2 | 379 |
| 54 | 4-NHCOH | 2 | 498 |
Selected compounds were submitted for evaluation of physicochemical properties and metabolic stability (Table 6). Typically, LogD and PSA values of all compounds were within targeted ranges, while solubilities were generally acceptable at low pH but barely acceptable or unacceptably low at physiological pH. Metabolic stability, expressed as an in vitro intrinsic clearance value, was determined by measuring the rate of degradation of test compound when incubated with human or mouse liver microsomes. The 3′-tetrazole compound 20 was observed to possess excellent metabolic stability - no measureable degradation of the compound was observed in both human and mouse liver microsomes (Table 6). Other compounds ranged widely in their metabolic stabilities, generally within acceptable limits except for ureas 44, 45 and 50, which were excessively labile in human microsomes. Intriguingly, 48 was markedly more stable in mouse microsomes than in human microsomes. Protein plasma binding was also measured in human plasma or was predicted using a chromatographic method employing a human serum albumin column and was well within the acceptable limit of 99.5% for all compounds where assessed.
| Solubility (μg/mL)a | LogDb (pH 7.4) | PSAc (Å) | In vitro CLint (μL/min/mg protein)d | PPB e (%) | ZR75-1 f IC50 (μM) | LIMK2 g IC50 (nM) | LIMK1 h IC50 (nM) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 6.5 | pH 2 | Mouse | Human | |||||||
| a Measured using nephelometry (see supplementary information†). b Measured chromatographically (see supplementary information†). c Calculated as topological PSA by ACL/Labs software, version 9.12. d Measured in liver microsomes (see supplementary information†). e If asterisked, estimated based on a chromatographic method using a human serum albumin column; otherwise, measured from human plasma viacentrifugation (see supplementary information†). f Inhibition of cofilin phosphorylation in ZR75-1 cell line (see supplementary information†). g Assayed as described in supplementary information.† h Values taken from Tables 2–4. i To be a candidate for lead optimization using internally defined selection criteria. j Could not calculate: no measurable degradation was observed. k Structures shown in Fig. 2. | ||||||||||
| Targeti (ideal/acceptable) | >100/>10 | >100/>10 | 1-4/-1-5 | <120/<140 | <10/<54 | <8/<42 | <99.5 | <1/<10 | ||
| Compound | ||||||||||
| 6 | <1.6 | 6.3–12.5 | 3.65 | 80 | 839 | 114 | — | — | — | 300 |
| 7 | 3.1–6.3 | 12.5–25 | 3.4 | 57 | 572 | 70 | — | — | — | 260 |
| 18 | 3.1–6.3 | 25–50 | 1.9 | 123 | 26 | c.n.c. (<5)j | 96.3* | >30 | 273 | 120 |
| 20 | 6.3–12.5 | 6.3–12.5 | 2 | 134 | c.n.c. (<6)j | c.n.c. (<5)j | 98.8* | >30 | 90 | 50 |
| 40 | 1.6–3.1 | 12.5–25 | 2.4 | 121 | 68 | 24 | — | >30 | 164 | 87 |
| 44 | 3.1–6.3 | 50–100 | 2.8 | 112 | 56 | 77 | — | 2.4 | 103 | 95 |
| 45 | 12.5–25 | >100 | 2.5 | 115 | 27 | 165 | 96.3 | 1.6 | 157 | 95 |
| 48 | 6.3–12.5 | 50–100 | 2.1 | 138 | c.n.c (<6)j | 68 | 94.7 | 1.4 | - | 81 |
| 50 | <1.6 | 50–100 | 2.7 | 122 | 111 | 116 | >98.6 | 4.1 | 133 | 90 |
| BMS3 k | 1.6–3.1 | 1.6–3.1 | 3.8 | 88 | 34 | 15 | 97.1 | 0.22 | 85 | 10 |
| BMS4 k | 1.6–3.1 | 1.6–3.1 | 3.0 | 143 | 10 | c.n.c. (<5)j | — | 1.0 | 696 | 54 |
Compared with parent compound 6, many of these properties are vastly improved. In particular, 6 is diabolically insoluble at pH 6.5 and above to the point of being unworkable, and was a reason this compound was not subjected to some of the other biological assays listed in Table 7. This compound was metabolically much less stable than all the other compounds in mouse liver microsomes, and much less stable than the better compounds such as 18, 20 and 40 in human liver microsomes. Its LogD was also much higher than other members in the series. Compound 7 fares better, as it is more soluble than 6 and more stable to human liver microsomes, but its LogD is high and it is very unstable to mouse liver microsomes. Compound 7 in many respects would appear to be a better platform for optimisation than 6, but as we have shown in Table 5, our early efforts to improve activity did not meet with success.
| Compound | CYP (% inhibition at 20 μM)a | hERG b | Relevant Off Target Kinases (% inhibition at 1 μM)c | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3A4 | 1A2 | 2C19 | 2D6 | 2C9 | JAK1 | JAK2 | JAK3 | Lck | Src | PAK4 | PAK1 | ROCK2 | ||
| a Assayed as described in the supplementary information.† b hERG fluorescence polarization assay performed by CEREP (% inhibition at 10 μM). c Assayed as described in the supplementary information.† d Could not calculate: no measurable inhibition was observed. | ||||||||||||||
| 44 | 82 | 28 | 53 | 9 | 75 | 0 | 13 | 95 | 56 | 71 | 60 | 7 | 0 | 10 |
| 45 | 82 | 11 | 48 | 12 | 32 | 27 | 25 | 88 | 53 | 75 | 71 | 0 | 33 | 24 |
| 48 | 70 | 17 | 14 | c.n.c.d | 16 | 0 | 31 | 97 | 65 | 97 | 45 | 0 | 23 | 18 |
| 50 | 99 | 13 | 69 | 74 | 62 | 11 | 31 | 98 | 60 | 85 | 23 | 13 | 8 | 13 |
Biomarker response was assessed by inhibition of cofilin phosphorylation in ZR75-1 cells and by this measure, compounds 44, 45, 48 and 50 were gratifyingly potent. However, compounds 18, 20 and 40 were inactive in this biomarker assay for reasons yet to be conclusively elucidated. Compounds 18 and 40 are the only amides that are either primary (18) or secondary (40) in this table while the tetrazole in 20 would be negatively charged. It is therefore possible that some combination of physicochemical properties (permeability, solubility, crystallinity) in the intracellular assay is not captured by the physicochemical data in Table 6 may be responsible for the lack of intracellular biomarker response. It is also possible that compounds 44, 45, 48 and 50 have some other off-target activity that is manifest in inhibition of intracellular cofilin phosphorylation. Further studies will be required to inform on this matter.
In order to gauge selectivity of compounds that were active in the biomarker assay, testing against an in-house kinase panel was undertaken that deliberately included the pathway-relevant kinases PAK1, PAK4 and ROCK2. As shown in Table 7, here only negligible inhibition was observed at a test concentration of 1 μM. However, significant inhibition of some other kinases, such as JAK3, Lck, Src and in particular JAK2 was observed at the test concentration of 1 μM. In order to assess the potential of this system for drug-drug interactions, this compound set was tested for inhibition of cytochrome P450 (CYP) isoforms using a test concentration of 20 μM. As shown in Table 7, CYP3A4 was strongly inhibited by all four compounds, particularly by amide 50. However, the 3-morpholino glycinamide 48 did not significantly inhibit any of the other CYP isoforms at the test concentration. This compound set was then evaluated for toxic pharmacology through inhibition of hERG. Pleasingly, none of the compounds in Table 7 exhibited significant hERG inhibition at a test concentration of 10 μM.
A comparison of our compounds was performed with some more advanced LIMK1/2 inhibitors reported by workers at Bristol-Myers-Squibb,4 namely BMS3 and BMS4 (Fig. 2). BMS3 and BMS46 were synthesised and their potent LIMK1/2 inhibitory activity4 was generally confirmed with our in-house LIMK1/2 assay. However, the LIMK2 activity of BMS4 was significantly different, returning an inhibitory IC50 value of 696 nM versus the reported 14 nM.4BMS3 and BMS4 were further assessed for their physicochemical and predictive ADME properties, as well as their biomarker inhibition in ZR75-1 cells. As listed in Table 6, BMS3 potently inhibited cellular phosphorylation of cofilin, with an EC50 of 220 nM, however BMS4 was significantly less potent, with an EC50 of 1 μM. BMS3 is exquisitely specific for LIMK1/2 over more than 200 other kinases4 and has good stability to human liver microsomes and can so be regarded as a more advanced tool compound than any currently in our series. We would seek to establish similarly stringent characteristics during lead optimisation of our series. Evenso, our early compounds already compare favourably with regard to a number of physicochemical aspects with BMS3 and BMS4. In particular, BMS3 has a significantly higher LogD of 3.8 and 3.0 respectively, while BMS4 has a LogD and polar surface area higher than any of our compounds. Furthermore, both are very poorly soluble at pH 2 and at pH 6.5. As such, the new class of LIMK1 inhibitors we report herein stand as promising platforms for optimization towards therapeutic relevance.
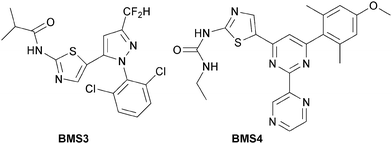 | ||
| Fig. 2 Structures of BMS3 and BMS4 as reported in ref. 4 as compounds 3 and 4 respectively. The IC50 values for BMS3 against LIMK1 and LIMK2 given in ref. 4 are 7 nM and 8 nM respectively, while we determined them to be 10 nM and 85 nM respectively as listed in Table 6. The IC50 values for BMS4 against LIMK1 and LIMK2 given in ref. 4 are 22 nM and 14 nM respectively, while we determined them to be 54 nM and 696 nM respectively as listed in Table 6. | ||
Conclusions
Early stage development of the 7-phenyl-4-aminobenzothieno[3,2-d]pyrimidines8 has seen the series move towards promising drug-like space with respect to potency, selectivity, distribution coefficient and polar surface area, solubility, plasma protein binding, microsomal stability, biomarker response, CYP inhibition and lack of hERG activity. This series of ATP-competitive inhibitors of LIMK1 now contains candidates suitable for lead optimization where many of these properties may be improved to furnish useful tool compounds as an alternative class with more favourable properties to those currently reported.3Acknowledgements
The authors acknowledge the financial support of the Cancer Therapeutics CRC, established and supported under the Australian Government's Cooperative Research Centres Program; NHMRC IRIISS grant number 361646 and Victorian State Government OIS grant; Diane Crighton and Elizabeth Trivier, from Cancer Research Technology Ltd, for providing ZR75-1 data.Notes and References
- R. W. Scott and M. F. Olson, J. Mol. Med., 2007, 85, 555–568 CrossRef CAS.
- O. Bernard, Int. J. Biochem. Cell Biol., 2007, 39, 1071–1076 CrossRef CAS.
- F. Manetti, Med. Res. Rev., 2011 DOI:10.1002/med.20230.
- P. Ross-Macdonald, H. de Silva, Q. Guo, H. Xiao, C.-Y. Hung, B. Penhallow, J. Markwalder, L. He, R. M. Attar, T.-a. Lin, S. Seitz, C. Tilford, J. Wardwell-Swanson and D. Jackson, Mol. Cancer Ther., 2008, 7, 3490–3498 CrossRef CAS.
- S. T. Wrobleski, S. Lin, K. Leftheris, L. He, S. P. Seitz, T.-a. Lin and W. Vaccaro, US Pat. Appl. 0178388.
- B. E. Sleebs, I. P. Street, X. Bu and J. B. Baell, Synthesis, 2010, 7, 1091–1096 CrossRef.
- H. A. Burgoon, N. C. Goodwin, B. A. Harrison, J. P. Healy, Y. Liu, R. Mabon, B. Marinelli, D. B. Rawlins, D. S. Rice and N. A. Whitlock, US Patent 2009264450 Chem Abstr 2009 151:491144 28–16.
- B. A. Harrison, S. D. Kimball, R. Mabon, D. B. Rawlins, D. S. Rice, M. V. Voronkov and Y. Zhang, WO 2009021169 CAN 150:237638 28–17.
- B. A. Harrison, N. A. Whitlock, M. V. Voronkov, Z. Y. Almstead, K.-J. Gu, R. Mabon, M. Gardyan, B. D. Hamman, J. Allen, S. Gopinathan, B. McKnight, M. Crist, Y. Zhang, Y. Liu, L. F. Courtney, B. Key, J. Zhou, N. Patel, P. W. Yates, Q. Liu, A. G. E. Wilson, S. D. Kimball, C. E. Crosson, D. S. Rice and D. B. Rawlins, J. Med. Chem., 2009, 52, 6515–6518 CrossRef CAS.
- B. E. Sleebs, A. Levit, I. P. Street, H. Falk, T. Hammonds, W.A.-C., M. D. Charles and J. B. Baell, Med. Chem. Commun., 2011 10.1039/c1md00137J.
Footnote |
| † Electronic supplementary information (ESI) available: Details of assay protocols, synthetic procedures and compound characterisation. See DOI: 10.1039/c1md00138h |
| This journal is © The Royal Society of Chemistry 2011 |