Design, synthesis, and structure–activity relationships of 1,3,4-oxadiazol-2(3H)-ones as novel FAAH inhibitors†
László E.
Kiss
a,
Humberto S.
Ferreira
a,
Alexandre
Beliaev
a,
Leonel
Torrão
b,
Maria João
Bonifácio
b and
David A.
Learmonth
*a
aLaboratory of Chemistry, Department of Research & Development, BIAL - Portela & Ca., S.A., S. Mamede do Coronado, Portugal. Fax: +351-22-9866192; E-mail: david.learmonth@bial.com; Tel: +351-22-9866100
bLaboratory of Pharmacology, Department of Research & Development, BIAL - Portela & Ca., S.A S, Mamede do Coronado, Portugal
First published on 3rd August 2011
Abstract
Novel 5-aryloxy substituted 3-phenyl-1,3,4-oxadiazol-2(3H)-ones were prepared and identified as potent inhibitors of FAAH. In vitro SAR are discussed. Structural variations of the selected lead compound were explored in order to optimise in vivo efficacy and selectivity.
Introduction
First synthesised in 1899,13-phenyl-5-methoxy-1,3,4-oxadiazol-2(3H)-one (1a) attracted little attention until the 1970's, when analogues such as 1b and 1c were shown to possess anthelmintic properties.2 Later, the 5-phenoxy derivatives 2a and 2b were demonstrated to have similar biological activity (Fig. 1).3 | ||
| Fig. 1 | ||
In the early 2000's, 5-alkoxy derivatives (e.g.3 and 4) were developed for the treatment of obesity and diabetes, based on their ability to inhibit pancreatic lipase (PL) and hormone-sensitive lipase (HSL).4 The 3-phenyl-5-methoxy-1,3,4-oxadiazol-2(3H)-one fragment also contributes a significant part of the structure of certain benzoylureas (e.g.5) reported for the treatment of type 2 diabetes (Fig. 2).5
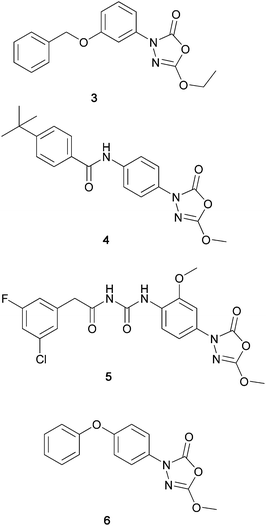 | ||
| Fig. 2 | ||
We envisaged that the 3-phenyl-1,3,4-oxadiazol-2(3H)-one moiety could also serve as a useful template for the design of improved fatty acid amide hydrolase (FAAH) inhibitors. FAAH, like PL and HSL, is a member of the extensive family of serine hydrolases6 and catalyses the degradation of a class of lipid signalling fatty acid amides, including oleamide and anandamide.7Anandamide elicits a range of biological responses with potential therapeutic relevance for the treatment of pain and inflammation amongst other disorders, and therefore FAAH has become recognised as a promising therapeutic target.8 Accordingly, numerous small molecule inhibitors belonging to various chemical classes have been reported (for a recent review see9 and references cited therein).
Our group submitted a patent application (with a priority date of December 2007) claiming a novel series of 3,5-disubstituted 1,3,4-oxadiazol-2(3H)-ones as potent FAAH inhibitors,10 derived from the initial screening hit 7 (3-(3-chlorophenyl)-5-methoxy-1,3,4-oxadiazol-2(3H)-one) (Fig. 3). A few weeks before this disclosure reached the public domain, FAAH inhibitory activity was reported for several HSL inhibitors, including seven 3-phenyl-1,3,4-oxadiazol-2(3H)-ones,11 with compound 6 (Fig. 2) clearly the most potent inhibitorin vitro with an IC50 of 6.1nM.
 | ||
| Fig. 3 | ||
Extremely limited structure–activity relationship (SAR) data was disclosed in this basic screening study, with the curious exception that replacement of the 5-methoxy group with a 5-phenoxy substituent resulted in a 10-fold decrease in potency in vitro. This is in stark contrast to findings within our own series. Furthermore, since only in vitro data was reported, no assessment of efficacy of FAAH inhibition in vivo could be established. Thus, we herein report the synthesis and explore the SAR of a distinct series of 3,5-disubstituted-1,3,4-oxadiazol-2(3H)-ones and reveal an unexpected highly selective preference for peripheral FAAH inhibition in vivo.
Chemistry
The synthetic route to obtain the necessary key intermediates from commercially available reagents is briefly outlined in Scheme 1.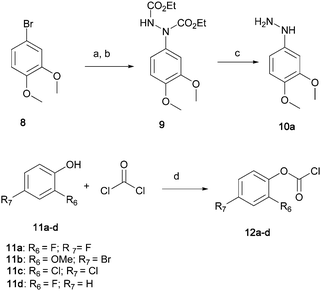 | ||
| Scheme 1 Reagents and conditions: (a) Mg, THF, cat. 1,2-dibromoethane, 80 °C. (b) DEAD, THF, −78 °C, then rt. (c) KOH, IPA-water, reflux (d).N, N-dimethylaniline, toluene, 0 °C. | ||
The relatively unstable 3,4-dimethoxy phenylhydrazine 10a was prepared as previously described12 by reaction of the Grignard reagent derived from 8 with diethylazodicarboxylate (DEAD) and subsequent hydrolysis of the intermediate 9 thereby obtained.
Commercially unavailable phenyl chloroformates were prepared by reacting appropriately substituted phenols 11a–d with phosgene in toluene in the presence of N, N-dimethylaniline13 to give 12a–d in excellent yield (85–95%). Construction of the oxadiazolone ring was accomplished via the general method depicted in Scheme 2. Hydrazines 10a–y, were acylated with selected acyl chlorides 12a–y in N-methylpyrrolidin-2-one (NMP) containing pyridine. Intermediates 13a–y thus obtained were treated with phosgene in dichloromethane at room temperature to give the target compounds.14
 | ||
| Scheme 2 (a) Pyridine, NMP, 0 °C, then rt. (b) Phosgene, DCM, 0 °C, then rt. | ||
Results and discussion
In vitro FAAH inhibition was determined in rat brain homogenates, using tritium (3H) labelled anandamide (AEA) by measuring the formation of 3H-ethanolamine as previously described.15 The results for a representative selection of new compounds are provided in Table 1. The initial screening hit 7 displayed fairly promising activity (95% inhibition at a concentration of 10 μM). The low molecular weight of 7, in conjunction with a fairly simple synthetic route, led us to consider this molecule as a suitable starting point for a hit-to-lead optimization programme. In the first phase, variation of the side chains (R11 and R22) by alternative groups was investigated.
Substitution of the methoxy group of 7 with –N-morpholinyl 14, phenyl 15, benzoyl 16, benzyl 17 or benzyloxy 18 residues completely abolished or drastically reduced FAAH inhibition.
However, it was then discovered that inhibition could be completely restored by incorporation of a 5-phenoxy group as in 19a. This immediately provided clarification that an O-substituent at position 5 of the oxadiazolone ring is absolutely essential for activity against FAAH.
Compound 19a was thus selected as an early lead for further optimization. Various 5-phenoxy-3-phenyl-1,3,4-oxadiazol-2(3H)-one derivatives were initially evaluated in vitro at two different concentrations (IC50 values were determined for compounds showing >50% inhibition at 0.1 μM). Whilst lead 19a exhibited 45% inhibition at a concentration of 0.1 μM, it was found to be completely inactive at a ten-fold lower concentration (Table 2).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | R3 | R4 | R5 | R6 | R7 | 0.1 μMa,c | 0.01 μMa,c | Liverb,c | Brainb,c | IC50 nMd |
| a % of Control. b 30 mg kg−1, po, FAAH activity was determined 1 h after administration. c Results are mean ± SEMs of four experiments. d IC50 values are presented with 95% of confidence intervals. | ||||||||||
| 19a | H | H | H | H | H | 56 ± 7 | 97 ± 9 | ND | ND | ND |
| 19b | H | H | H | H | –OCH3 | 7 ± 1 | 61 ± 2 | 94 ± 4 | 108 ± 6 | 16 (11; 21) |
| 19c | H | H | H | H | –OH | 80 ± 14 | ND | ND | ND | ND |
| 19d | H | H | H | H | Cl | 6 ± 2 | 31 ± 4 | 81 ± 9 | 110 ± 1 | 5 (4; 5) |
| 19e | H | H | H | F | F | 1 ± 0 | 14 ± 1 | 39 ± 10 | 94 ± 13 | 3 (2; 4) |
| 19f | H | H | H | –OCH3 | Br | 1 ± 0 | 16 ± 1 | 83 ± 6 | 101 ± 9 | 4 (3; 6) |
| 19g | −CN | H | H | H | H | 96 ± 3 | ND | ND | ND | ND |
| 19h | H | –NO2 | H | H | H | 78 ± 5 | 103 ± 10 | ND | ND | ND |
| 19i | H | H | –OCH3 | H | H | 112 ± 19 | ND | ND | ND | ND |
| 19j | −OH | H | H | H | H | 28 ± 3 | 91 ± 8 | 46 ± 15 | 97 ± 6 | 52 (43; 63) |
| 19k | H | −OH | H | H | H | 14 ± 3 | 90 ± 15 | 33 ± 7 | 95 ± 2 | 32 (21; 46) |
| 19l | H | H | F | H | H | 3 ± 0 | 43 ± 8 | 72 ± 8 | 97 ± 2 | 7 (5; 10) |
| 19m | F | H | F | H | H | 3 ± 0 | 46 ± 2 | 65 ± 10 | 86 ± 2 | 9 (6; 12) |
| 19n | −OH | H | H | H | –NO2 | 0 ± 0 | 5 ± 1 | 45 ± 16 | 96 ± 4 | 2 (1; 2) |
| 19o | −OH | H | H | H | Cl | 0 ± 0 | 6 ± 1 | 20 ± 5 | 38 ± 13 | 3 (2; 3) |
| 19p | −OH | H | H | Cl | Cl | 0 ± 0 | 5 ± 1 | 9 ± 8 | 59 ± 31 | 1 (1; 2) |
| 19q | −OH | H | H | F | H | 1 ± 0 | 53 ± 12 | 44 ± 18 | 94 ± 6 | 10 (8; 13) |
| 19r | −OH | H | H | H | F | 2 ± 0 | 51 ± 5 | 30 ± 2 | 82 ± 8 | 14 (10; 20) |
| 19s | −OH | H | H | F | F | 0 ± 0 | 4 ± 0 | 12 ± 3 | 86 ± 11 | 2 (2; 2) |
| 19t | −OH | H | F | H | Cl | 0 ± 0 | 5 ± 1 | 4 ± 1 | 64 ± 23 | 1 (1; 2) |
| 19u | H | –OH | H | H | Cl | 1 ± 0 | 25 ± 3 | 10 ± 4 | 100 ± 7 | 4 (2; 6) |
| 19v | H | −OH | H | H | F | 3 ± 0 | 57 ± 12 | 24 ± 2 | 99 ± 6 | 11 (6; 18) |
| 19w | H | –OH | H | F | F | 0 ± 0 | 10 ± 2 | 12 ± 2 | 94 ± 5 | 2 (2; 4) |
| 19x | –OH | –OH | H | F | F | 0 ± 0 | 25 ± 3 | 4 ± 1 | 66 ± 6 | 4 (2; 7) |
| 19y | –OH | –OH | H | H | Cl | 0 ± 0 | 34 ± 5 | 5 ± 1 | 83 ± 39 | 6 (3; 10) |

The effects on FAAH inhibitory activity of different substituents on the 3-phenyl- and 5-phenoxy rings of the 1,3,4-oxadiazol-2(3H)-one nucleus are shown in Table 2. The 4-methoxy derivative 19b displayed increased inhibition over the parent 19a, whereas removal of the methyl group led to the inactive phenol 19c.
The inclusion of halogen atoms was found to be favourable. The 4-chloro derivative 19d displayed promising inhibition (69% at 0.1 μM) which was surpassed by the fluoro-analogue 19e with the 4-bromo-2-methoxy analogue 19f essentially equipotent. On the other hand, introduction of strongly electron-withdrawing substituents (19g, cyano and 19h, nitro) on the “west-side” of the molecule appeared to be less favourable. A methoxy group at the ortho position of the 3-phenyl ring (19i) was not tolerated at all. However, the presence of a hydroxyl group at either the para- (19j) or meta position (19k) resulted in an approximately two to three fold increase in FAAH inhibition compared to 19a. The presence of fluorine at either the ortho- (19l) or para (19m) positions was also found to be beneficial in terms of potency.
All compounds for which IC50 values were determined were then subsequently assessed for in vivoFAAH inhibition. Compounds 19b, 19d–f and 19j–k were administrated orally to mice at a dose of 30 mg/kg. Thereafter, at 1 h post-administration, the animals were sacrificed and FAAH activity was determined in liver and brain. As seen in Table 2, despite the promising in vitro activity of most of these compounds (19b, 19d–f and 19l–m), only the difluoro analogue 19e showed evidence of in vivo efficacy. Somewhat surprisingly, the at least ten-fold less active hydroxy compounds (19j–k) in vitro showed good in vivo inhibition with a clear preference for peripheral FAAH. A hydroxyl group in either the meta- or para position was found to be essential for high inhibition, indicating that this substituent is likely to be involved in binding to the protein. Furthermore, the presence of this polar, hydrogen bond donating group should decrease the overall lipophilicity of the compounds whilst simultaneously increasing aqueous solubility. These characteristics could contrive to restrict permeation of the molecules across the blood-brain barrier, resulting in selective peripheral FAAH inhibition. Thus, subsequent efforts focussed on this hydroxyl series (19j–k), leading to the elaboration of derivatives 19n–y shown in the lower half of Table 2. From the in vitro SAR, introduction of electron-withdrawing substituents on the 5-phenoxy ring was found to be clearly beneficial, such that all of these compounds presented IC50 values in the low nanomolar range. In parallel with the in vitro assay, the ability of compounds 19n–y to inhibit FAAH in mice was investigated. The para-nitro substituted derivative 19n was found to be equipotent to the parent hydroxy compound 19j. Replacement of the nitro group with chlorine as in 19o gave a compound that demonstrated an approximately two-fold improvement in potency for liver FAAH but which also performed reasonably well in the brain. Incorporation of a second chlorine atom (19p) provided a further increase in liver FAAH inhibition with more enhanced selectivity. Conversely, the ortho-fluoro analogue 19q failed to provide enhanced inhibition over the parent compound 19j, although the para–fluoro isomer 19r exhibited slightly higher activity. Combining the substitution patterns of compounds 19q and 19r resulted in the 2,4-difluoro derivative 19s, which was equipotent to the dichloro analogue 19p in the periphery with no traceable effect in the brain. As mentioned earlier, the introduction of a fluorine atom in the ortho position of the 3-phenyl ring (19l) provided a dramatic increase in in vitro potency. Based on this finding, we were prompted to synthesise the corresponding 3-(2-fluorophenyl) analogue of 19o. Gratifyingly, the resulting compound 19t exhibited five-fold greater efficacy over the parent 19o with considerably improved peripheral selectivity.
For completeness, a few examples of meta-hydroxy substituted derivatives (19u–w) were prepared for in vivo comparison to their para-hydroxy counterparts (19o and 19r–s). Compounds 19u–w were generally endowed with more enhanced liver FAAH inhibition over 19o and 19r–s, and furthermore they were found to be exquisitely peripherally selective inhibitors of FAAH. Thereafter, we were encouraged to consider introducing a further hydroxyl group to these molecules. Although the more lipophilic difluoro catechol 19x demonstrated some degree of central inhibition, the mono-chloro catechol 19y exhibited potent, highly selective peripheral FAAH inhibition.
Finally, we examined the time-dependent FAAH inhibition by those compounds that provided over 90% in vivoFAAH inhibition. Table 3 details the differences in the FAAH inhibitory profiles of compounds 19p, 19t and 19x–y in mouse liver and brain homogenates.
Compounds 19p and 19t were found to achieve maximum inhibitory effect in the liver at 1 h after oral administration at a dose of 30 mg kg−1. Thereafter they sustained constant inhibition of FAAH over the next 7 h, followed by a gradual return to baseline levels at 24 h post-administration. The extent of central FAAH inhibition by oxadiazolones 19p and 19t was markedly lower compared to peripheral inhibition and hovered around the 50% level up to 8 h post-dose. Catechols 19x–y were orally administered to mice at a lower dose of 10 mg/kg. Both compounds achieved 90–95% FAAH inhibition in the liver within 1 h post-dose and had no measurable effect (Table 2) in the brain at any timepoint. This finding may be of therapeutic relevance, as the purely peripheral selective nature of FAAH inhibition by compounds 19x–y could serve to reduce potential side effects caused by inhibition of FAAH in the central nervous system.
Conclusion
Novel substituted 5-phenoxy-3-phenyl-1,3,4-oxadiazol-2(3H)-ones 19a–m derived from the initial in vitro screening hit 7 were found to be potent in vitroinhibitors of FAAH. Compounds 19j–k displayed promising in vivo inhibition over compounds 19b, 19d, 19f and 19l–m. Consequently, an optimised series of 3-(hydroxyphenyl)-5-phenoxy-1,3,4-oxadiazol-2(3H)-ones 19n–y were prepared and found to possess enhanced in vivoFAAH inhibition over the parent phenols 19j–k. Catechols 19x–y showed potent and peripherally selective in vivoFAAH inhibition at a dose of 10 mg kg−1 and may be beneficial in the treatment of certain cardiovascular disorders such as hypertension and heart failure. Compounds 19n–y are currently being further evaluated for their therapeutic potential.Experimental section
Chemistry
The purity of test compounds in all cases was higher than 95%. Analytical TLC was performed on precoated silica gel plates (Merck 60 Kieselgel F 254) and visualized with UV light. Solvents and reagents were purchased from Aldrich, Merck, and Fluka and were used without further purification.3,4-Dimethoxy phenylhydrazine 10a
To a stirred mixture of 4-bromo-1,2-dimethoxybenzene 8 (10 g, 46.1 mmol) and magnesium turnings (1.232 g, 50.7 mmol) in THF (185 mL) was added 1,2-dibromoethane (0.20 g, 1.06 mmol). The reaction was heated at reflux for 2 h, and then cooled to −78 °C, whereupon diethyl diazene-1,2-dicarboxylate (8.02 g, 7.25 mL, 46.1 mmol) was added dropwise. After stirring for 15 min in the cold, followed by 2 h at room temperature, acetic acid (0.6 g) was added followed by water. The mixture was extracted with ethyl acetate and the organic phase was dried over MgSO4, then filtered and evaporated. Chromatography (petroleum ether-ethyl acetate, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) followed by trituration in a mixture of diethyl ether-petroleum ether gave the title compound 9 as a beige powder (10.4 g, 72%).
:1) followed by trituration in a mixture of diethyl ether-petroleum ether gave the title compound 9 as a beige powder (10.4 g, 72%).
General procedure for the synthesis of oxadiazolones 14–18, 19a–b and 19d–i, 19l–m
Pharmacology
Frozen brains (without cerebellum) from Wistar rats were used, and each brain was homogenized in 15 ml 1 mM MgCl2, 20nM HEPES pH 7.0 with Potter Elvejhem (8 strokes at 500 rpm). Homogenates were centrifuged for 20 min at 36000g at 4 °C (Beckman, 70Ti rotor). Pellets were resuspended in 15 ml of the same buffer and centrifuged under the same conditions. Pellets were resuspended in 15 ml of the same buffer and incubated for 15 min at 37 °C after which they were centrifuged for 20 min at 36000g at 4 °C. Each pellet was then resuspended in 15 ml 3 mM MgCl2, 1 mM EDTA, 50 mM Tris pH 7.4 and protein determined with BioRad Protein Assay (BioRad) using a standard curve of BSA (50–250 μg ml−1). The membrane suspensions were aliquoted and stored at −80 °C.
The FAAH activity was determined using AEA (labelled with 3H in the ethanolamine part of the molecule) as substrate and measuring the 3H-ethanolamine formed. Reaction mix (total volume of 200 μl) contained: 2 μM AEA (2 μM AEA + 5 nM 3H-AEA), 0.1% fatty acid free BSA, 5 μg protein, in 1 mM EDTA, 10 mM Tris pH 7.6 and 10 μM or 100 mM compounds. Stock solutions of the compounds to test (10mM) were prepared in 100% DMSO and the DMSO concentration in the assay was 0.1%. After a 15 min preincubation period at 37 °C, reaction was started by the addition of the substrate solution (cold EAE + radiolabelled EAE + BSA). Reaction was carried out for 10 min before termination by the addition of 400 μl activated charcoal suspension (8 g charcoal in 32 ml 0.5 M HCI in continuous agitation). After a 30 min incubation period at room temperature with agitation, charcoal was sedimented by centrifugation in microfuge (10 min at 13000 rpm). 200 μl of the supernatant were added to 800 μl Optiphase Supermix scintillation cocktail previously distributed in 24-well plates. Counts per minute (cpm) were determined in Microbeta TriLux scintillation counter (10 min counting or until σ = 2). In each assay blanks (no protein, usually below 200 cpm) and controls (no compound) were present. The results are reported as % of control after blank subtraction.
Animal treatment. The animals used for experiments were male NMRI mice (weighing 27–44 g) obtained from Interfauna Ibérica (Spain). Mice were kept 5 per cage, under controlled environmental conditions (12 h light/dark cycle and room temperature 22 ± 1 °C). Food and tap water were allowed ad libitum and the experiments were all carried out during daylight hours.
Animals were administered 30 mg kg−1 test compounds via oral route (8 ml kg−1; compound suspended in 0.5% carboxymethylcellulose (CMC) or solubilized in water) or 8ml kg−1 0.5% CMC (controls) using animal feeding stainless steel curve needles (Perfectum, USA). Fifteen minutes before sacrifice animals were anesthetised with pentobarbital 60 mg kg−1 administered intraperitoneally. A fragment of liver and brain without cerebellum were removed and put in plastic vials containing membrane buffer (3 mM MgCl2, 1 mM EDTA, 50 mM Tris HCl pH 7.4). Tissues were stored at −30 °C until analysis. Animals were fasted overnight before administration of compounds except for time periods of >18h, where food was removed on the morning of the day of administration and the compound was administered in the afternoon of the same day. Animals were then given water but nothing else.
All animal procedures were conducted in the strict adherence to the European Directive for Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (86/609CEE) and Portuguese legislation (Decreto-Lei 129/92, Portarias 1005/92 e 1131/97). The number of animals used was the minimum possible in compliance with current regulations and scientific integrity.
Reagents and solutions. Anandamide [ethanolamine -1-3H-] (40–60Ci/mmol) was obtained from American Radiochemicals. All other reagents were obtained from Sigma-Aldrich. Optiphase Supermix was obtained from Perkin Elmer and activated charcoal was obtained from Sigma-Aldrich.
Tissue preparation. Tissues were thawed on ice and were homogenised in 10 volumes of membrane buffer (3 mM MgCl2, 1 mM EDTA, 50 mM Tris HCl pH 7.4) with either Potter-Elvejhem (brains - 8 strokes at 500 rpm) or Heidolph Diax (livers - 2 strokes at position 5 for 20 s with 30 s pauses). Total protein in tissues was determined with the BioRad Protein Assay (BioRad) using a standard curve of BSA (50–250 μg ml−1).
Enzymatic assay. Reaction mix (total volume of 200 μl) contained: 2 μM AEA (2 μM AEA + 5 nM 3H-AEA), 0.1% fatty acid free BSA, 15 μg (brain), 5 μg (liver) or 50 μg (lung) protein, in 1 mM EDTA, 10 mM Tris pH 7.6. After a 15 min pre-incubation period at 37 °C, reaction was started by the addition of the substrate solution (cold AEA + radiolabelled AEA + BSA). Reaction was carried out for 10 min (brain and lung) or 7 min (liver) before termination by the addition of 400 μl activated charcoal suspension (8 g charcoal in 32 ml 0.5 M HCl in continuous agitation). After a 30 min incubation period at room temperature with agitation, charcoal was sedimented by centrifugation in microfuge (10 min at 13000 rpm). 200 μl of the supernatant were added to 800 μl Optiphase Supermix scintillation cocktail previously distributed in 24-well plates. Counts per minute (cpm) were determined in a MicrobetaTriLux scintillation counter.
In each assay blanks (without protein) were prepared. The percentage of remaining enzymatic activity was calculated with respect to controls (no compound) and after blank subtraction.
References
- S. Busch, J. Prakt. Chem., 1899, 60, 239 Search PubMed; H. Busch, Chem. Ber., 1901, 34, 2332 Search PubMed.
- R. Boesch (Rhone-Poulenc Industries), GB Patent 1518042, 1976.
- G. Bonse, N. Müller and A. Harder(Bayer AG), European Patent EP0419918A2 1990.
- K. Schoenafinger, S. Petry, G. Mueller and K.-H. Baringhaus(Aventis Pharma Deutschland GmbH), US Patent 6369088B2, 2002; K. Schoenafinger, S. Petry, G. Mueller, A. Bauer and H. O. Heuer(Aventis Pharmaceuticals Inc), US Patent 0236288A1, 2003.
- K. Schoenafinger, E. Defossa, D. Kadereit, E. Von Roedern, T. Klabunde, H.-J. Burger, A. Herling and K.-U. Wendt(Sanofi-Aventis Deutschland GmbH), US Patent 7138414B2, 2006.
- M. K. McKinney and B. F. Cravatt, Annu. Rev. Biochem., 2005, 74, 411 CrossRef CAS.
- B. F. Cravatt, D. K. Giang, S. P. Mayfield, D. L. Boger, R. A. Lerner and N. B. Gilula, Nature, 1996, 384, 83 CrossRef CAS.
- B. F. Cravatt and A. H. Lichtman, Curr. Opin. Chem. Biol., 2003, 7, 469 CrossRef CAS.
- H. Deng, Expert Opin. Drug Discovery, 2010, 5, 961 CrossRef CAS.
- D. A. Learmonth, L. E. Kiss, A. Beliaev, H. Ferreira and P. Soares da Silva(BIAL-Portela & Companhia S.A.), PCT Int. Appl. WO2009084970A1, 2009.
- A. Minkkilä, J. R. Savinainen, H. Käsnänen, H. Xhaard, T. Nevalainen, J. T. Laitinen, A. Poso, J. Leppänen and S. M. Saario, Chem. Med. Chem., 2009, 4, 1253 CrossRef.
- J. P. Demers (Ortho Pharmaceuticals Co.) US Patent 4864032, 1989.
- F. Strain, W. E. Bissinger, W. R. Dial, H. Rudoff, B. J. DeWitt, H. C. Stevens and J. H. Langstone, J. Am. Chem. Soc., 1950, 72, 1254 CrossRef CAS.
- J. Huang, D. F. Bushey and M. D. Graves, J. Heterocycl. Chem., 1987, 24, 1 CrossRef CAS.
- S. Holt, F. Comelli, B. Costa and C. J. Fowler, Br. J. Pharmacol., 2005, 146, 467 CrossRef CAS; L. Boldrup, S. J. Wilson, A. J. Barbier and C. J. Fowler, J. Biochem. Biophys. Methods, 2004, 62, 171 CrossRef; M. Steffens, A. Schulze-Bonhage, R. Surges and T. J. Feuerstein, Neurosci. Lett., 2005, 385, 13 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and elemental analysis data. See DOI: 10.1039/c1md00136a |
| This journal is © The Royal Society of Chemistry 2011 |