Structure–activity relationship of conformationally constrained peptidomimetics for antiproliferative activity in HER2-overexpressing breast cancer cell lines†
Sashikanth
Banappagari
,
Sharon
Ronald
and
Seetharama D.
Satyanarayanajois
*
Department of Basic Pharmaceutical Sciences, College of Pharmacy, University of Louisiana at Monroe, 1800 Bienville Drive, Monroe, LA 71201, USA. E-mail: jois@ulm.edu; Fax: +1-(318)-342-1737; Tel: +1-(318)-342-1993
First published on 17th June 2011
Abstract
Human epidermal growth factor receptor 2 (HER2) is a member of the human epidermal growth factor receptor kinases and is involved in a signaling cascade for cell growth and differentiation. It is well established that HER2-mediated heterodimerization has important implications in cancer. Deregulation of signaling pathways and overexpression of HER2 are known to occur in cancer cells, indicating the role of HER2 in tumorigenesis. Therefore, blocking HER2-mediated signaling has potential therapeutic value. We have designed several peptidomimetics to inhibit HER2-mediated signaling for cell growth. One of the compounds (compound 5, Arg-[3-amino-3(1-naphthyl)-propionic acid]-Phe) exhibited antiproliferative activity with IC50 values in the nanomolar to micromolar range in breast cancer cell lines. To further investigate the structure–activity relationship of the compounds, various analogs of compound 5 were designed. Conformational constraints were initiated in the peptidomimetic with introduction of a Pro residue in the peptidomimetic sequence. Results of antiproliferative activity indicated that analogs of compound 5 with C-and N-terminal ends capped compound 16 and compound 9 with Asp at the C-terminal exhibited antiproliferative activity in the lower micromolar range against breast cancer cell lines. Introduction of conformational constraints such as Pro residue in the sequence or cyclization did not enhance the activity of the peptidomimetic. Competitive binding studies were carried out to evaluate the binding of potent peptidomimetics to HER2-overexpressing cancer cell lines. Results indicated that compounds exhibiting antiproliferative activity in breast cancer cell lines bind to the cells that overexpress HER2 protein.
1. Introduction
Epidermal growth factor receptors (EGFR), a growth factor receptor family of proteins that control normal cell growth, differentiation, and motility have been widely studied.1,2 When extracellular ligands such as epidermal growth factor (EGF) bind to the extracellular ligand binding domain of EGFR, receptor homo-heterodimerization, activation of tyrosine kinase activity, and autophosphorylation of the receptors results, thus initiating a mitogenic signaling cascade.3–8 Human epidermal growth factor receptor 2 (HER2) is a member of human epidermal growth factor receptor kinases that are involved in a signaling cascade for cell growth and differentiation. It is well established that HER2-mediated heterodimerization has important implications in cancer.9–13 Overexpression of HER2 and its heterodimerization with other epidermal growth factor receptors are known to occur in cancer cells.14–17When HER2-mediated heterodimerization is obstructed, the result is inhibition of phosphorylation, which ultimately leads to control of cell growth. Thus, blocking HER2-mediated signaling has significant therapeutic potential. Monoclonal antibodies specifically directed against the extracellular domain of the HER2 have been shown to selectively inhibit the growth of HER2-overexpressing cancer cells.13,18 The antibody herceptin (trastuzumab) binds to the extracellular domain (ECD) IV of HER2 and inhibits the cleavage site of MMPs.18–20 There are several reports of the design of molecules directed against HER2 protein as promising alternatives to traditional non-specific chemotherapy.9,21–25 Peptidomimetics and peptide vaccines have been reported to interfere with the dimer interface of HER2 with EGFR.9,26,27 At present no small molecule specifically targeting an extracellular region of HER2 has been approved for clinical use. Therefore, there is a need to develop small molecules that are targeted toward the extracellular region of the HER2. In an earlier report we have described the design, synthesis, docking studies, and antiproliferative activity of peptidomimetics based on the crystal structure of HER2 complexed with its antibody herceptin (Fig. 1).28 We have also shown that a peptidomimetic compound 5 exhibits antiproliferative activity in the lower micromolar range and specifically binds to HER2-overexpressing breast cancer cells.29 To further investigate the structure–activity relationship of compound 5 and its analogs, we investigated the inclusion of different functional groups (amino acids or β-amino acids) in the peptidomimetic and studied their antiproliferative activity in breast cancer cell lines. Conformational constraints were introduced by incorporating a Pro residue in the peptidomimetic sequence. Compound 5 is a peptidomimetic with N- and C-termini that are viable for enzymatic degradationin vivo. It was modified by conformational constraints in the structure to give various analogs using criteria such as N- and C-terminal modifications and cyclization of the main chain. The N-terminal of 5 was acetylated and the C-terminal was amidated. The peptide was also stabilized by backbone cyclization and cyclization with introduction of the dibenzofuran (DBF) functional group.30 Peptidomimetics produced by such modifications are shown in Tables 1 and 2. CellTiter-Glo or MTT assays were performed on BT-474, MCF-7, and SKBR-3 cell lines in the presence of peptidomimetics to evaluate the anti-proliferative activity against breast cancer cell lines. Among the designed peptidomimetic analogs of 5, compounds 16 and 9 exhibited antiproliferative activity against HER2-overexpressing breast cancer cell lines in the lower micromolar range. Furthermore, competitive binding studies using fluorescently labeled 5 suggested that compound 9 specifically binds to HER2-overexpressing breast cancer cells.
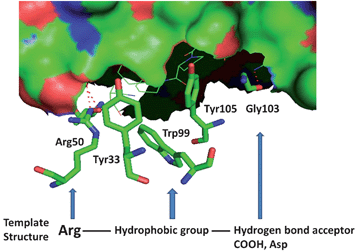 | ||
| Fig. 1 Structure of domain IV of HER2 with antibody herceptin. HER2 is shown in surface. Amino acid residues from herceptin that interact with domain IV of HER2 are shown. The template structure for the peptidomimetic design is also shown at the bottom of the figure. | ||
| Code number | Compound | BT-474 IC50/μM | MCF-7 IC50/μM |
|---|---|---|---|
| a For lapatinib IC50 values reported are from lit. 35. MCF-7 cell lines do not respond to lapatinib. | |||
| 5 |
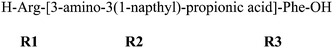
|
0.895 ± 0.029 | 16.9 ± 1.0 |
| 6(SS) | H-Arg-[3-amino-3(1-naphthyl)-propionic acid]-Tyr-OH | 46 | >50 |
| 6(RR) | H-Arg-[3-amino-3(1-naphthyl)-propionic acid]-Tyr-OH | >50 | >50 |
| 7(SS) | H-Arg-[3-amino-3(1-naphthyl)-propionic acid]-Asp-OH | >50 | >50 |
| 7(RR) | H-Arg-[3-amino-3(1-naphthyl)-propionic acid]-Asp-OH | >50 | >50 |
| 8 | H-Arg-[3-amino-3(1-naphthyl)-propionic acid]-[3-amino-3(1-naphthyl)-propionic acid]-OH | >50 | >50 |
| 9 | H-Arg-[3-amino-3(1-naphthyl)-propionic acid]-Phe-Asp-OH | 0.785 ± 0.011 | 45 |
| 10(SS) | H-Arg-[3-amino-3(biphenyl)-propionic acid]-Phe-OH | 5.8 ± 0.2 | >50 |
| 10(RR) | H-Arg-[3-amino-3(biphenyl)-propionic acid]-Phe-OH | 2.46 ± 0.35 | >50 |
| 11(SS) | H-Lys-[3-amino-3(biphenyl)-propionic acid]-Phe-OH | >50 | >50 |
| 11(RR) | H-Lys-[3-amino-3(biphenyl)-propionic acid]-Phe-OH | >50 | >50 |
| 12(SS) | H-Arg-Pro-[3-amino-3(1-naphthyl)-propionic acid]-Phe-OH | 1.95 ± 0.14 | 40 |
| 12(RR) | H-Arg-Pro-[3-amino-3(1-naphthyl)-propionic acid]-Phe-OH | 1.96 ± 0.16 | >50 |
| 13(SS) | H-Arg-Pro-[3-amino-3(1-naphthyl)-propionic acid]-Tyr-OH | >50 | >50 |
| 13(RR) | H-Arg-Pro-[3-amino-3(1-naphthyl)-propionic acid]-Tyr-OH | >50 | >50 |
| Lapatinib | 0.025 ± 0.004 | N.A |
2. Results
Antiproliferative activity of the peptidomimetics
To evaluate the anticancer activity of the designed compounds, synthesized compounds were screened for antiproliferative activity using the microculture tetrazolium assay (MTT), or CellTiter-Glo assay.31–33 The MTT assay is based on the ability of metabolically active cells to reduce the yellow tetrazolium salt to a colored formazan product, whereas the CellTiter-Glo assay is a method of determining the number of viable cells based on quantitation of ATP present.33 The IC50 values for the various compounds are shown in Tables 1 and 2. For comparison, IC50 value for lapatinib, a dual kinase inhibitor of EGFR and HER2, is also provided.34,35 Note that lapatinib binds to the kinase domain of HER2 (and EGFR) which is different from the extracellular domain. The lead compound 5 was shown to be highly selective for HER2-overexpressing cell lines, with an IC50 value of 0.396 μM in SKBR-3 and 0.896 μM in BT-474 cell lines and 16.9 μM in MCF-7 cell lines.28,29 To evaluate the effect of R1 and R3 flanking residues of the central β-amino acid, peptidomimetics with different functional groups at R1 and R3 were evaluated (Table 1). Replacement of the C-terminal amino acid Phe with Tyr (a hydrophobic group with a hydroxyl group) (compound 6) or a negatively charged acidic side chain (compound 7) resulted in a loss of activity. Replacement of the N-terminal positively charged amino acid Arg with a similar amino acid Lys also resulted in a loss of activity of the compounds (11). Introduction of a β-amino acid with a hydrophobic side chain at the R3 position also resulted in a loss of activity (8). Introduction of a bulky hydrophobic group in the central portion (R2) of the molecule (10) retained the activity and specificity of the peptidomimetics for HER2-overexpressing breast cancer cell lines. To understand the relative positions of Arg-β-amino acid-Phe and the spacing between them, a Pro residue was introduced, resulting in compounds 12 and 13. Pro is also known to induce stability in the peptide structure with a β-turn type of conformation.36 Compound 12 retained the antiproliferative activity with an IC50 value of 1.95 μM. When compound 12 was modified at the C-terminal with Tyr, activity was lost (13). These results suggest that N-terminal Arg and C-terminal Phe are important for the activity of the peptidomimetic and that the central residue of the peptidomimetic should have a bulky hydrophobic group such as naphthalene or biphenyl ring structure. To stabilize the structure against enzymatic degradation and to attain secondary structure such as β-turn in the peptide, a cyclization strategy was used (14 and 15). Cyclization of the peptidomimetic with a Pro residue resulted in a moderate loss of activity of the compound. However, when the compound was cyclized with a β-turn/β-sheet-inducing organic linker DBF, the activity was completely lost (15). This clearly suggests that the peptidomimetic may not need secondary structure such as a β-turn to bind to its receptor. In order to stabilize the N- and C-termini of the peptide, 5 was end-capped with an acetyl group and an amide group. The resulting peptidomimetic (16) exhibited moderate antiproliferative activity. A Cys amino acid was introduced (17) in the compound for conjugation of fluorescent and other molecules in later studies. Introduction of Cys at the C-terminal resulted in low antiproliferative activity. Variation in the chirality of the β-amino acid was also investigated. Compounds with R or S configurations at the central β-amino acids did not show any significant variation in activity (example, 6(SS), 6(RR), 10(SS), 10(RR)). Overall, the modifications of the peptidomimetic with different amino acids flanking the central β-amino acids or introduction of conformational constraints such as cyclization seemed to decrease the activity of the peptide. To understand the importance of the distance between the N-terminal positive charge, the central hydrophobic residue, and the C-terminal Phe group with a free carboxy terminal (Fig. 1), compound 9 with Asp at the C-terminal was designed. Introduction of an Asp residue (9) at the C-terminal resulted in enhanced activity of the compound compared to that of the parent compound (5). Selectivity of the compound for HER2-overexpressing cancer cell lines was retained, as shown by the IC50 value of 0.785 μM for compound 9 in BT-474 cell lines that overexpress HER2 protein and 45 μM in MCF-7 cell lines that do not overexpress HER2 protein.| Code no. | Structure | BT-474 IC50/μM | SKBR-3 IC50/μM | MCF-7 IC50/μM |
|---|---|---|---|---|
| a N.A not applicable. | ||||
| 14 |
|
7.79 ± 0.77 | 6.38 ± 0.68 | 17.22 ± 1.85 |
| 15 |
|
>50 | >50 | >50 |
| 16(SS) |
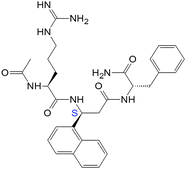
|
1.02 ± 0.25 | 1.40 ± 0.45 | 11.78 ± 1.43 |
| 16(RR) |
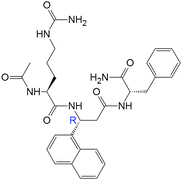
|
6.37 ± 0.31 | 9.75 ± 0.33 | 26.13 ± 1.75 |
| 17(SS) |
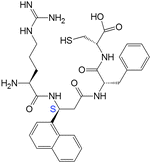
|
11.07 ± 1.74 | 12.46 ± 1.54 | 28.43 ± 1.87 |
| 17(RR) |
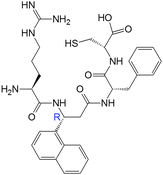
|
13.7 ± 1.6 | 15.63 ± 1.59 | 31.32 ± 1.25 |
| FITC-5 |
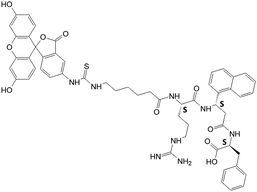
|
N.A | N.A | N.A |
Competitive binding studies
In an earlier report we have shown that compound 5 specifically binds to HER2-overexpressing breast cancer cell lines.29 Compounds 16 and 9 showed antiproliferative activity in the lower micromolar range. N-and -C-termini end-capping of compound 16 makes it stable against enzymatic degradationin vivo.37,38 However, we wanted to investigate whether the modification still retained the specificity of binding to HER2-overexpressing cancer cell lines. The objective of this aim is to evaluate the binding of 16 and 9 (Tables 1 and 2) to HER2 on the cell surface using fluorescently labeled 5 (FITC-5) (Table 2). A competitive binding assay was carried out to investigate the ability of unlabeled 16 and 9 to inhibit the binding of FITC-5 on BT-474 cells. Fig. 2 shows the competitive binding assay results. A comparison of the binding of 50 μM FITC-5 and 16 or 9 (Fig. 2A and B) clearly suggests that unlabeled 16 and 9 compete with FITC-5 to bind with HER2 protein on BT-474 cell lines. Similar results were obtained with SKBR-3 cell lines. Compound 15, which does not exhibit potent antiproliferative activity against cancer cell lines, was used as a control (Table 2 and Fig. 2C). Compound 15 does not bind competitively to HER2-overexpressing cancer cell lines, suggesting that 16 and 9 bind specifically to HER2-overexpressing cell lines.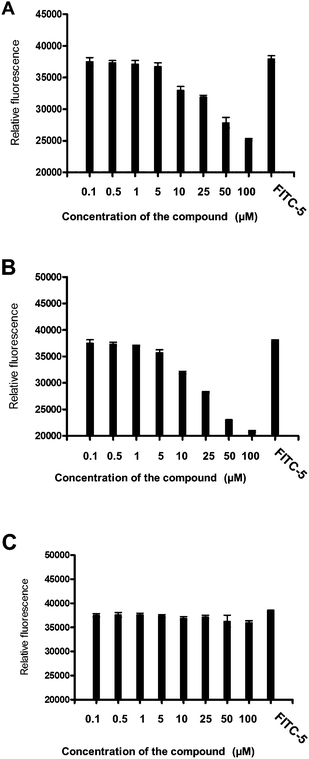 | ||
| Fig. 2 Competitive binding of compounds with FITC-labeled compound 5 at different concentrations to BT-474 cells that overexpress HER2 protein. Relative fluorescence intensity was represented. FITC-5 concentration was 50 μM. Concentration of unlabeled peptide is shown on the X-axis. (A) Compound 16, (B) compound 9, and (C) compound 15. Notice that compounds 16 and 9 replace the binding of FITC-5 at a concentration of ≤5 μM. | ||
3. Discussion
Design strategy
The crystal structure of HER2–herceptin complex indicates that the binding site on the HER2 domain IV has a pocket-like structure (Fig. 1) homologous to that of domain II of other HERs.5 This pocket accommodates binding of small peptide/peptidomimetic molecules that can modulate HER2-mediated signaling. Domains II and IV of the HER2 extracellular region play major roles in multimerization of HERs and the downstream signaling that leads to cell growth.6 Herceptin (anti-HER2) binds to HER2 on the C-terminal part of domain IV. This interaction is known to block HER2 from proteolytic cleavage and indirectly affect the dimerization with other HERs that induce signaling pathways. Blocking domain IV of HER2 with antibodies is known to suppress the growth of cancer cells by indirectly inhibiting the dimerization mechanism of HER2 with other receptors. It is also known that the C-terminal portions of the domains interact with one another in EGFR and HER2, and that the absence of domain IV of HER2 changes the heteromeric signaling of EGFR.20Peptides designed from the C-terminal of domain IV with a disulfide loop have been shown to inhibit EGFR–HER2 interaction.5 Thus, blocking any interaction at these regions on HER2 can disrupt signaling pathway(s) that promote cell proliferation and cancer. We designed a template compound based on the spatial disposition of electrostatic and hydrophobic interaction sites in the HER2–herceptin complex. The designed template is shown in Fig. 1 along with HER2–antibody herceptin interactions.5 Using a rational drug design approach, we designed and synthesized (a peptidomimetic) compound 5 (Table 1) that showed antiproliferative activity against HER2-overexpressing breast cancer cell lines with IC50 of 0.396 μM and 16.9 μM in MCF-7 cell lines.28 This specificity of compound 5 toward HER2-overexpressing breast cancer cell lines prompted us to modify the peptidomimetic to investigate the structure–activity relationship of 5. To understand the importance of functional groups in compound 5, the location of positively charged Arg and hydrophobic groups and C-terminal hydrogen bond acceptor groups and the effect of these groups on the antiproliferative activity of the designed compounds, several analogs of compound 5 were designed. Positively charged Arg was labeled R1, central β-amino acid was labeled R2, and the C-terminal hydrophobic group was labeled R3 for the purpose of design (Table 1). To evaluate the effect of R1 and R3 flanking residues of the central β-amino acid, peptidomimetics with different functional groups at R1 and R3 were assessed, resulting in compounds 6–8. The C-terminal Phe was changed to Tyr, a hydrophobic amino acid with a hydroxyl functional group (6). The C-terminal hydrophobic group was replaced with a negatively charged acidic amino acid Asp, resulting in 7. Compound 8 was designed to evaluate the effect of bulky hydrophobic groups at R3. The hydrophobic functional group of the β-amino acid was changed to a bulky biphenyl group, resulting in compounds 10 and 11. In compound 11, along with a central β-amino acid, the N-terminal Arg was replaced with a similar amino acid Lys. Furthermore, conformational constraints were introduced in the peptide by introducing a Pro residue in the sequence. The amino acid Pro with an imino group is known to restrict the conformation of small peptides. The Pro residue is also known to introduce β-turn structure in the peptides.36,39–42 Such β-turn structures have been shown to enhance biological activity by restricting the conformation of the peptide. Pro-Pro and Pro-Gly sequences have been shown to form β-turn structures in proteins and peptides.43 A Pro residue placed in the second position in the β-turn seems to have a high propensity for forming a turn.44 Hence, a Pro residue was introduced in the second position in the sequence, resulting in compounds 12 and 13. The amino acids in the peptidomimetic had S (L) chirality. However, the β-amino acid can have R and S chirality. To investigate the effect of chirality of the β-amino acid on the conformation and biological activity, we designed diastereoisomers of peptidomimetics with both R and S configurations at βNaph (for example, 6(SS), 6(RR)).Antiproliferative activity
To restrain the conformation of the molecule for enhanced activity, cyclic versions of the compounds were designed with backbone cyclization (14, 15) (note that cyclization of three amino acid residue peptides is not possible unless a linker or a functional group is added to the peptidomimetic). However, cyclization did not enhance the activity of the compounds. To increase the stability of the compounds in the cellular environment,37,38N- and C-termini of 5 were capped, producing compound 16. The antiproliferative activity of compound 16 was slightly decreased by N- and C-terminal modification as indicated by the IC50 value of 1.96 μM. To further investigate the effect of additional functional groups on compound 5, compounds 8 and 17 were designed. Compound 9 was designed based on the fact that, in the crystal structure of the HER2–herceptin complex (Fig. 1), the three intermolecular interactions are hydrogen bonding between Arg50 of Herceptin and Asp560 as well as Glu558 of HER2, hydrophobic interaction of Phe573 of HER2 with Tyr33 and Trp99 of herceptin, and hydrogen bond interaction between Gly103 of herceptin with Lys593 of HER2. The hydrogen bonding interaction of Gly103 occurs farther away from Arg50 and the hydrophobic interaction (Tyr33, Trp99 and Tyr105). Thus, if we introduce an Asp with a negatively charged side chain next to the Phe in the peptidomimetic design, the side chain of Asp should be at an appropriate distance from Arg and the hydrophobic group to mimic the carbonyl carbon of Gly103 hydrogen bonding in the crystal structure (Fig. 1). Compound 9 exhibits IC50 values of 0.785 μM in HER2-overexpressing BT-474 cell lines and 45 μM in MCF-7 cell lines that do not overexpress HER2 protein; this makes it the most potent compound among the analogs of compound 5 studied. Furthermore, competitive binding studies using fluorescently labeled 5 suggested that compound 9 specifically binds to HER2-overexpressing breast cancer cells (Fig. 2B).From our structure–activity studies, it is suggested that an Arg at the N-terminal, a central hydrophobic/aromatic group, or an acidic group or a hydrogen-bonding acceptor (such as an Asp side chain) in the C-terminal of the peptidomimetic is essential for the activity. Introduction of the conformationally constrained residue Pro in the peptidomimetic sequence (12) did not enhance the activity of the peptidomimetic. Furthermore, compared to the parent compound 5, 12 has one additional residue Pro, and this additional amino acid may change the orientation of pharmacophore groups in the peptidomimetic to the receptor compared to compound 5.
4. Materials and methods
Peptidomimetics
Peptidomimetics were designed and custom synthesized by NeoMPS (San Diego, CA), Aroztech LLC (Cincinnati, OH) and New England Peptide (Gardner, MA). The purity of the peptides was confirmed by HPLC and the identity of the correct molecular ion was confirmed by mass-spectrometry. For synthesis of compounds 5, 8, 9, 14, 15 and FITC-5, β-amino acid used had S chirality. For other compounds described in this report, β-amino acid used was a racemic mixture (use of chirally pure β-amino acid was very expensive as many analogs of compound 5 were designed and synthesized). During purification by HPLC, the epimers could be separated. Chirality of the epimers was identified by CD spectra. For reference CD spectra of compound 5 was used. Compound 5 was synthesized by solid phase peptide synthesis using standard Fmoc amino acids.45,46 FITC-5 was synthesized using Fmoc-Phe-Wang resin. Acp (Fmoc-6-aminocaproic acid) was linked to the peptide on the resin followed by FITC.47 Cleavage and deprotection with TFA gave crude peptide. Compound was purified by HPLC (HPLC chromatogram, mass spectrum and CD spectra of compounds and synthesis scheme for FITC-5 are provided as ESI†).MTT and CellTiter-Glo assays for the determination of antiproliferative activity
The growth inhibitory activity of target compounds was determined on cell lines BT-474, SKBR-3 (human breast cancer cell line which overexpresses HER2), and MCF-7 (human breast cancer cell line that do not overexpress HER2), using a modified version of the microculture tetrazolium assay.31,32BT-474 and MCF-7 cells were maintained in RPMI 1640 supplemented with 10% FBS and 1% antibiotics; cells of passages 5 onward were used, and BT-474 cell lines were maintained according to the supplier's guidelines (ATCC, Manassas, VA). SKBR-3 cell lines were maintained in McCoy's modified medium.Once the cells reached 90% confluency, a cell suspension was prepared by trypsinization of monolayer cultures. Cell counts were performed, and the suspensions were diluted accordingly to give 1 × 104cells per mL with the appropriate medium. Aliquots (100 μL) of the cell suspension were added to each well in a 96-well microtitre plate. The cells were incubated for 24 h (37 °C, 5% CO2). Stock solutions of the test compound were prepared in DMSO, and serial dilutions were made with medium to over a hundredfold concentration range. Not more than 1% DMSO (final concentration) was present in each well. The test sample was incubated with the cells for 72 h. Wells without cells and those with cells in culture medium/DMSO were examined in parallel. 0.1% SDS was included as a positive control. At the end of the incubation period, the medium was decanted and replaced with 100 μL MTT solution (0.5 mg mL−1 in 1× phosphate buffer saline solution (PBS). The cells were incubated for another 3 h, after which the medium was removed from each well by pipetting and the cells were carefully washed with PBS (100 μL). DMSO (150 μL) was added to each well to lyse the cells and dissolve the purple formazan crystals. The absorbance of the formazan product was measured within 30 min at 590 nm on a microtitre plate reader.
The CellTiter Glo33,48 assay kit was obtained from Promega Corporation (Madison, WI). After the compounds were incubated with cells for 72 h, the wells were washed with PBS and 100 μL CellTiter-Glo reagent was added to cells containing 100 μL medium. The plates were equilibrated for 20 min and luminescence values from the cells with and without the compounds were read using blank cells with 0.1% SDS and 1% DMSO.
The absorbance/luminescence values obtained at each concentration (triplicates for each run and 3–4 independent runs were carried out) were averaged, adjusted by subtraction of blank values (wells without cells), and expressed as a percentage of the average absorbance obtained from control wells (in the absence of test compound). IC50 values were determined from logarithmic plots of the % absorbance versus concentration generated using GraphPad Prism (San Diego, CA). The reported IC50 values were calculated from 3–4 independent experiments.
Competitive binding assay with unlabeled HERP5 peptide
BT-474/MCF-7 cells were plated at a density of 104cells per well in a 96-well tissue culture plate and incubated at 37 °C with 5% CO2 for 24 h. Compounds 16, 15, and 9 were diluted in PBS at different concentrations. A mixture of FITC-5 and different concentrations of 16, 15, or 8 were prepared. In each well, a mixture of FITC-5 and the test compound was added, keeping the concentration of FITC-5 constant. The plates were incubated for 45 min at 37 °C with 5% CO2 After washing, fluorescence from the cells was read in a microplate reader using an excitation wavelength of 485 nm and an emission wavelength of 528 nm. Autofluorescence values from cells were subtracted and the fluorescence from the cells was expressed as relative fluorescence from each sample. A plot of relative fluorescence vs. concentration of the compound was plotted.5. Conclusions
Peptidomimetics with conformational constraints were designed based on the HER2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :herceptin crystal structure. The peptidomimetics designed exhibited antiproliferative activity against breast cancer cell lines. Introduction of conformational constraints such as a Pro residue and cyclization of the peptidomimetic did not enhance the activity of the peptidomimetic compared to that of the parent compound 5. A positively charged Arg at the N-termini, a hydrophobic group with a constrained side chain, and a properly placed negatively charged residue at the C-termini seem to enhance the antiproliferative activity of the peptidomimetic (compound 9). The structure–activity results presented here will help us to design small molecules that have high specificity toward HER2-overexpressing breast cancer cell lines. Replacement of Phe with Trp amino acid at the C-terminal of the peptidomimetic and binding of these molecules to the extracellular domain of the HER2 protein are in progress to further understand the interactions of analogs of compound 5 with HER2 protein.
:herceptin crystal structure. The peptidomimetics designed exhibited antiproliferative activity against breast cancer cell lines. Introduction of conformational constraints such as a Pro residue and cyclization of the peptidomimetic did not enhance the activity of the peptidomimetic compared to that of the parent compound 5. A positively charged Arg at the N-termini, a hydrophobic group with a constrained side chain, and a properly placed negatively charged residue at the C-termini seem to enhance the antiproliferative activity of the peptidomimetic (compound 9). The structure–activity results presented here will help us to design small molecules that have high specificity toward HER2-overexpressing breast cancer cell lines. Replacement of Phe with Trp amino acid at the C-terminal of the peptidomimetic and binding of these molecules to the extracellular domain of the HER2 protein are in progress to further understand the interactions of analogs of compound 5 with HER2 protein.
Acknowledgements
The project described was supported by grant number P20RR016456 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. Authors would like to thank the Mass Spectrometry Facility and NMR facility, Department of Chemistry, Louisiana State University Baton Rouge for high resolution mass spectra of compounds.References
- N. E. Hynes and H. A. Lane, Nat. Rev. Cancer, 2005, 5, 341–354 CrossRef CAS.
- J. Baselga and S. M. Swain, Nat. Rev. Cancer, 2009, 9, 463–475 CrossRef CAS.
- J. R. Woodburn, Pharmacol. Ther., 1999, 82, 241–250 Search PubMed.
- S. Hobbs, E. M. Cameron, R. P. Hammer, A. T. D. Le, R. M. Gallo, E. N. Blommel, S. L. Coffing, H. Chang, J. David and D. J. Riese, II, Oncogene, 2004, 23, 883–893 Search PubMed.
- H. S. Cho, K. Mason, K. X. Ramyar, A. M. Stanley, S. B. Gabelli, D. W. Denney and D. J. Leahy, Nature, 2003, 421, 756–760 CrossRef CAS.
- A. W. Burgess, H. S. Cho, C. Eigenbrot, K. M. Ferguson, T. P. Garrett, D. J. Leahy, M. A. Lemmon, M. X. Silwkowski, C. W. Ward and S. Yokoyama, Mol. Cell, 2003, 12, 541–552 CrossRef CAS.
- Y. Yarden, J. Baselga and D. Miles, Semin. Oncol., 2004, 31, 6–13 Search PubMed.
- M. Cristofanilli and G. N. Hortobagyi, Endocr.-Relat. Cancer, 2002, 9, 249–266 Search PubMed.
- A. Berezov, J. Chen, Q. Liu, H. T. Zhang, M. I. Greene and R. Murali, J. Biol. Chem., 2002, 277, 28330–28339 Search PubMed.
- M. A. Cobleigh, C. L. Vogel, D. Tripathy, N. J. Robert, S. Scholl, L. Fehrenbacher, J. M. Wolter, V. Paton, S. Shak, G. Lieberma and D. J. Slamon, J. Clin. Oncol., 1999, 17, 2639–2648 CAS.
- D. B. Agus, R. W. Akita, W. D. Fox, G. D. Lewis, B. Higgins, P. I. Pisacane, J. A. Lofgren, C. Tindell, D. P. Evans, K. Maiese, H. I. Scher and M. X. Sliwkowski, Cancer Cell, 2002, 2, 127–137 Search PubMed.
- K. M. Ferguson, Annu. Rev. Biophys., 2008, 37, 353–373 Search PubMed.
- J. Mendelsohn and J. Baselga, Oncogene, 2000, 19, 6550–6565 CrossRef CAS.
- R. Nahta and F. J. Esteva, Clin. Cancer Res., 2003, 9, 5078–5084 Search PubMed.
- S. Menard, S. M. Pupa, M. Campiglio and E. Tagliabue, Oncogene, 2003, 22, 6570–6578 Search PubMed.
- R. Perez-Soler, Oncologist, 2004, 9, 58–67 Search PubMed.
- D. J. Slamon, B. Leyland-Jones, S. Shak, H. Fuchs, V. Paton, A. Bajmonde, T. Fleming, W. Eiermann, J. Wolter, M. Pegram, J. Baselga and L. N. Norton, N. Engl. J. Med., 2001, 344, 783–792 CrossRef CAS.
- D. S. Leonard, A. D. K. Hill, L. Kelly, B. Dijkstra, E. McDermott and N. J. O'Higgins, Br. J. Surg., 2002, 89, 262–271 Search PubMed.
- M. A. Molina, J. Codony-Servat, J. Albanell, F. Rojo, J. Arribas and J. Baselga, Cancer Res., 2001, 61, 4744–4749 Search PubMed.
- M. C. Franklin, K. D. Carey, F. F. Vajdos, D. J. Leahy, A. M. De Vos and M. X. Sliwkowski, Cancer Cell, 2004, 5, 317–328 CrossRef CAS.
- S. Kamath and J. K. Buolamwini, J. Med. Chem., 2003, 46, 4657–4668 Search PubMed.
- S. K. Rabindran, C. M. Discafani, E. C. Rosfjord, M. Baxter, M. B. Floyd, J. Golas, W. A. Hallett, B. D. Johnson, R. Nilakantan, E. Overbeek, M. F. Reich, R. Shen, X. Shi, H. R. Tsou, Y. F. Wang and A. Wissner, Cancer Res., 2004, 64, 3958–3965 CrossRef CAS.
- B. E. Fink, G. D. Vite, H. Mastalerz, J. F. Kadow, S. H. Kim, K. J. Leavitt, K. Du, D. Crews, T. Mitt, T. W. Wong, J. T. Hunt, D. M. Vyas and J. S. Tokarski, Bioorg. Med. Chem. Lett., 2005, 15, 4774–4779 Search PubMed.
- A. Wissner, E. Overbeek, M. F. Reich, M. B. Floyd, B. D. Johnson, N. Mamuya, E. C. Rosfjord, C. Discafani, R. Davis, X. Shi, S. K. Rabindran, B. C. Gruber, F. Ye, W. A. Hallet, R. Nilakantan, R. Shen, Y. F. Wang, L. M. Greenberger and H. R. Tsou, J. Med. Chem., 2003, 46, 49–63 Search PubMed.
- S. L. Moulder, F. M. Yakes, S. K. Muthuswamy, R. Bianco, J. F. Simpson and C. L. Arteaga, Cancer Res., 2001, 61, 8887–8895 Search PubMed.
- N. K. Dakappagari, K. D. Lunte, S. Rawale, J. T. Steele, S. D. Allen, G. Phillips, R. T. Reilly and T. P. Kaumaya, J. Biol. Chem., 2005, 280, 54–63 Search PubMed.
- S. Allen, J. T. Garrett, S. V. Rawale, A. L. Jones, G. Philips, G. Forni, J. C. Morris, R. G. Oshima and T. P. Kaumaya, J. Immunol., 2007, 179, 472–482 Search PubMed.
- S. D. Satyanarayanajois, S. Villalba, L. Jianchao and G. M. Lin, Chem. Biol. Drug Des., 2009, 74, 246–257 Search PubMed.
- S. Banappagari, S. Ronald and S. D. Satyanarayanajois, J. Biomol. Struct. Dyn., 2010, 3, 289–308 Search PubMed.
- K. H. Mayo, R. P. Dings, C. Flader, I. Nesmelova, B. Hargittai, D. W. van der Schaft, L. I. van Eijk, D. Walek, J. Haseman, T. R. Hoye and A. W. Griffioen, J. Biol. Chem., 2003, 278, 45746–45752 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 5, 55–63 Search PubMed.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Huresy, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
- S. P. M. Crouch, J. Immunol. Methods, 1993, 160, 81–88 CrossRef CAS.
- H.-P. Kim, Y.-K. Yoon, J.-W. Kim, S.-W. Han, H.-S. Hur, J. Park, J.-H. Lee, D.-Y. Oh, S.-A. Im, Y.-J. Bang and T.-Y. Kim, PLoS One, 2009, 4, e5933 Search PubMed.
- P. S. Hegde, D. Rusnak, M. Bertiaux, K. Alligood, J. Strum, R. Gagnon and T. M. Gilmer, Mol. Cancer Ther., 2007, 6, 1629–1640 Search PubMed.
- R. Rai, S. Raghothama and P. Balaram, J. Am. Chem. Soc., 2006, 128, 2675–2681 CrossRef CAS.
- H. John, E. Maronde, W. G. Forssmann, M. Meyer and K. Adermann, Eur. J. Med. Res., 2008, 13, 73–78 Search PubMed.
- T. L. Nguyen, J. K. Chau, N. A. Perry, L. de Boer, S. A. J. Zaat and H. J. Vogel, PLoS One, 2010, 5, e12684 Search PubMed.
- F. J. Blanco, M. A. Jimenez, J. Herranz, M. Rico, J. Santoro and J. L. Nieto, J. Am. Chem. Soc., 1993, 115, 5887–5888 CrossRef CAS.
- K. S. Kee and S. D. Jois, Curr. Pharm. Des., 2003, 15, 1209–1224 Search PubMed.
- K. R. Acharya, E. D. Sturrock, J. F. Riordan and M. R. W. Ehlers, Nat. Rev. Drug Discovery, 2003, 2, 891–897 CrossRef CAS.
- V. J. Hruby, Nat. Rev. Drug Discovery, 2002, 1, 847–858 CrossRef CAS.
- E. De Alba, M. Rico and M. A. Jimenez, Protein Sci., 1999, 8, 2234–2244 CAS.
- E. G. Hutchinson and J. Thronton, Protein Sci., 1994, 3, 2207–2216 CrossRef CAS.
- Handbook of Combinatorial and Solid Phase Organic Chemistry: a Guide to Principles, Products and Protocols, Advanced Chemtech, Louisville, KY, 1998, pp. 329–372 Search PubMed.
- P. Lloyd-Williams, F. Albericio and E. Giralt, Chemical Approaches to the Synthesis of Peptides and Proteins, CRC, Boca Raton, Florida, 1997, pp. 19–82 Search PubMed.
- M. Jullian, A. Hernandez, A. Maurras, K. Puget, M. Amblard, J. Martinez and G. Subra, Tetrahedron Lett., 2009, 50, 260–263 CrossRef CAS.
- T. Riss, R. Moravec and A. Niles, Cell Notes, 2005, 16–21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC, mass spectrum and NMR data for peptides. See DOI: 10.1039/c1md00126d |
| This journal is © The Royal Society of Chemistry 2011 |