Potent and selective inhibitors of human peptidylglycine α-amidating monooxygenase†
Feihua
Cao
,
Allan B.
Gamble
,
Hye-Kyung
Kim
,
Hideki
Onagi
,
Mary J.
Gresser
,
Jamie
Kerr‡
and
Christopher J.
Easton
*
ARC Centre of Excellence for Free Radical Chemistry and Biotechnology, Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia. E-mail: easton@rsc.anu.edu.au
First published on 21st June 2011
Abstract
Fatty acid glycolates are nanomolar inhibitors of small cell lung carcinoma peptidylglycine α-amidating monooxygenase, with binding affinities orders of magnitude stronger than those of the analogous thioesters, the corresponding substrate glycine derivatives or the natural substrate procalcitonin.
Peptidylglycine α-amidating monooxygenase (PAM) is a bifunctional enzyme, which catalyses the final step in the biosynthesis of C-terminal peptide amides, being oxidative cleavage of their corresponding glycine-extended precursors (Scheme 1).1–5 The peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidylamidoglycolate lyase (PAL) subunits respectively induce the formation and reaction of the intermediate α-hydroxylated glycine derivatives. Amidated peptides produced in this manner include a wide variety of mammalian peptide hormones and have been implicated in a broad range of pathological conditions, such as asthma,6 inflammation7 and cancer.8PAM is also responsible for the biosynthesis of non-peptide amides including fatty acid amides such as oleamide, which affects sleep regulation.9 The important role of PAM combined with the fact that amidated peptide hormones are often thousands of times more active than their glycine-extended precursors has led to significant interest in its regulation.10–25
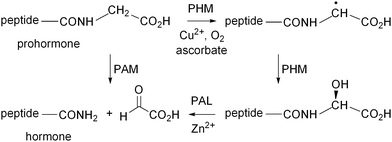 | ||
| Scheme 1 Biosynthesis of C-terminal amidated peptides catalysed by peptidylglycine α-amidating monooxygenase (PAM). | ||
trans-4-Phenylbut-3-enoic acid is an irreversible, mechanism-based inhibitor of PAM.10–12 It is reported to have a KI value of 96 nM against PAM of prostate cancer cells13 and is effective in vivo in reducing serum PAM activity14 as well as inducing antiinflammatory and analgesic effects.15,16 Other compounds of this class show at best micromolar activity as PAM substrates and inhibitors,17 as is the case with several other compound types,18–22 and only homocysteine derivatives that act by binding to active site copper in the PHM subunit are known to show potency down to 10 nM.23 A variety of substrate analogues having a glycolate instead of a glycine are reported to have KI or IC50 values26 against PAM ranging from 40–580 μM.24,25 Of these, the fatty acid derivative 1a was found to be marginally the most potent, prompting us to use it as the basis for further structure–activity-relationship studies with other fatty acid glycolates as well as with thioesters.
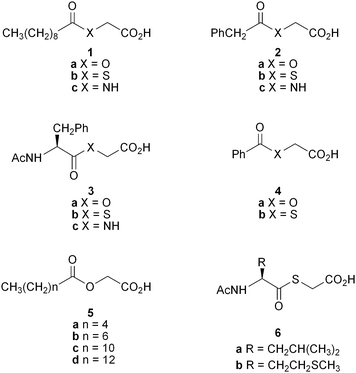
Our earlier work25 had used only Xenopus laevis (frog) PAM but in this study we also evaluated the compounds using PAM secreted into the media and extracted from cultured human cancer cell lines. We were specifically interested in identifying more potent and selective PAM inhibitors, more potent as they would be expected to be physiologically active at lower doses, and more selective for sub-types of PAM associated with particular disease states, so as to avoid complications associated with generic inhibition of PAM. Accordingly, we focused on human cancer cell lines where high levels of PAM have been reported. We now report that the glycolate 1a inhibits the enzyme extracted from H889 small cell lung carcinoma cells with an IC50 value of 50 nM. This represents an increase in binding affinity of almost three orders of magnitude over that seen for the glycolate 1a with frog PAM, whereas very little increase is seen with either the corresponding thioester 1b or substrate 1c. The binding affinity of the glycolate 1a (IC50 50 nM) with H889 PAM is also 2–3 orders of magnitude greater than that of the corresponding thioester 1b (IC50 7 μM) and substrate 1c (KM,app 200 μM), and the glycine-extended precursor of the natural growth hormone calcitonin (KM,app 300 μM). In addition, the glycolate 1a shows much greater potency against PAM extracted from mammalian cells than that secreted into the cell media. Similar patterns of reactivity are seen with the glycolates 2a, 3a and 5c, the thioesters 2b and 3b, and the glycine derivatives 2c and 3c, establishing that glycolates are peculiarly potent and selective inhibitors of human cancer cellular PAM.
The glycolates 1a and 4a had been prepared by treatment of benzyl bromoacetate with decanoic and benzoic acid, respectively, to give the corresponding benzyl esters that were then hydrogenated.25 The glycolates 2a, 3a and 5a–d were obtained using analogous procedures. Coupling mercaptoacetic acid and glycine directly under basic conditions with a range of carboxylic acids produced the thioglycolates 1b–4b and 6a–b, and the enzyme substrates 1c–3c, while glycine-extended calcitonin (procalcitonin) was purchased from GL Biochem (Shanghai) Ltd. Frog PAM was obtained from Wako Pure Chemical Industries. H889 and DMS53 small cell lung carcinoma, PC3 prostate cancer and SW1783 brain tumour cells were obtained from the American Type Culture Collection (ATCC). PAM activity has been reported previously for the H889, PC3 and SW1783 cell strains.27 The PC3 and H889 cell extracts are each reported to contain three alternatively spliced variants of the bifunctional form of PAM having both the PHM and PAL catalytic domains. Such data are not available for the DMS53 and SW1783 strains. PAM activity in the cultured cell media and extracts was assayed using the tripeptide substrate (R)-Tyr-(S)-Val-Gly, and the KM,app and IC50 values of the substrates 1c–3c, procalcitonin, and the enzyme inhibitors 1a,b–4a,b, 5a–d and 6a–b were then measured for various forms of PAM in competitive assays with the tripeptide. Details of the competitive assays and extraction procedures are provided in the ESI. The results are summarised in Tables 1 and 2.
| Glycolates IC50 (μM) | Thioesters IC50 (μM) | Glycine derivatives KM,app (μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| a Although KI, IC50 and KM,app values are different parameters, they are suitable for comparison as approximate measures of enzyme binding affinity, as used in the present work. The enzyme data are derived from experiments performed at least in duplicate and analysed at least in duplicate, where the results of the duplicate experiments and assays varied by less than 20%. b Data from ref. 25. | ||||||||
| Frog | H889 | Frog | H889 | Frog | H889 | |||
| 1a | 40b | 0.05 | 1b | 9 | 7 | 1c | 440 | 200 |
| 2a | 510 | 2 | 2b | 20 | 45 | 2c | 440 | 330 |
| 3a | >2000 | 6 | 3b | 25 | 45 | 3c | 170 | 15 |
| 4a | 250b | 4b | 150 | |||||
| 5a | 780 | 6a | 260 | |||||
| 5b | 250 | 6b | 50 | |||||
| 5c | 35 | 0.06 | ||||||
| 5d | 30 | |||||||
| Enzyme source | Glycolate 1a IC50 (μM) | Glycine derivative 1cKM,app (μM) | Procalcitonin KM,app (μM) |
|---|---|---|---|
| a See footnote a, Table 1. | |||
| Frog | 40 | 440 | |
| H889 cell extract | 0.05 | 200 | 300 |
| DMS53 cell extract | 0.04 | 30 | 200 |
| DMS53 cell medium | 30 | 110 | 360 |
| PC3 cell extract | 0.12 | ||
| SW1783 cell medium | 30 |
The glycolates 1a–4a and 5a–d have IC50 values with frog PAM ranging from 30 to >2000 μM (Table 1). The activity of the fatty acid derivatives 1a and 5a–d increases with increasing chain length, with the decanoate 1a (IC50 40 μM), the dodecanoate 5c (IC50 35 μM) and the tetradecanoate 5d (IC50 30 μM) showing the highest binding affinities of all the glycolates 1a–4a and 5a–d. The thioglycolates 1b–4b and 6a–b are also micromolar inhibitors of frog PAM, with compounds 1b–4b being 2 to >80 times more potent that the corresponding glycolates 1a–4a. The decanoate 1b (IC50 9 μM) shows the highest binding affinity of all the thioglycolates 1b–4b and 6a–b.
Interestingly, comparison of the IC50 values of the glycolate 1a (40 μM) and the thioglycolate 1b (9 μM) with the KM,app value of the corresponding glycine derivative 1c (440 μM) indicates that substitution of the substrate NH with either O or S has a positive rather than negative effect on binding to this enzyme (Table 1). The increased potency of the glycolate 1a is even more marked with PAM extracted from H889 human lung cancer cells, where the corresponding values for compounds 1a–c are 50 nM, and 7 and 200 μM. That is, the glycolate 1a and the thioglycolate 1b respectively bind 4,000 and 30 times more strongly than the glycine derivative 1c. This is somewhat anomalous based on a cursory consideration of the crystal structure of rat PHMcc,2 the catalytic core of the PHM subunit of PAM, with a bound peptide substrate. The substrate's glycine NH is hydrogen bonded to the carbonyl oxygen of the side chain of N316 of the protein, which would be expected to stabilise the enzyme-substrate complex relative to complexes of glycolates and thioglycolates where such an interaction would be lacking. However, the authors of the crystallographic report note that this enzyme-substrate hydrogen bond is only formed at the expense of breaking a hydrogen bond between protein amino acid residues Y318 and N316, and rotation of the N316 side chain, on substrate binding, so the binding energetics are difficult to predict. It is also possible that glycolates and thioglycolates could interact favourably with an asparagine side chain, though as hydrogen bond acceptors rather than donors. In any event, the interactions that determine the binding affinities of substrates and inhibitors obviously vary between enzyme from different sources, as the IC50 value of the glycolate 1a with PAM extracted from the H889 cells is 800 times lower than that with the frog enzyme, whereas no such decrease is seen with either the thioglycolate 1b or the glycine derivative 1c. Irrespective of the reasons, with an IC50 value of 50 nM, the glycolate 1a is a potent and effective inhibitor of the H889 PAM, given that the KM,app values of the corresponding glycine derivative 1c (200 μM) and the glycine-extended precursor of calcitonin (300 μM) are higher by 4,000 and 6,000 times, respectively (Table 2).
The trends observed with the decanoic acid derivatives 1a−c are also seen with other series of glycolates, thioesters and glycine derivatives. For example, the glycolates 2a and 3a have IC50 values with the H889 PAM which are >250 times lower than those for the frog enzyme. Against the H889 enzyme they also exhibit IC50 values 23 and 8 times lower than those of the respective thioesters 2b and 3b, and 165 and 3 times lower than the corresponding KM,app values of the analogous substrates 2c and 3c. The dodecanoate 5c is also a potent and selective inhibitor of the H889 PAM with an IC50 value of 60 nM.
The DMS53 lung cancer cells secrete active PAM into the cell medium. The glycolate 1a is much more potent as an inhibitor of the DMS53 cellular PAM (IC50 40 nM) than the secreted enzyme (IC50 30 μM, Table 2). PAM is known to undergo proteolytic processing while being transported from cells28 so different values are not unexpected but the reasons for the difference and the potent inhibition of the cellular enzyme are unclear. Although less directly comparable, the potency of the glycolate 1a as an inhibitor of PAM extracted from PC3 prostate cancer cells (IC50 120 nM) is also greater than that against the enzyme present in the medium of growing SW1783 brain tumour cells (IC50 30 μM). Calcitonin has been detected in extracts from DMS53 cells and, in this context, it is particularly noteworthy that the binding affinity of DMS53 cellular PAM with the glycolate 1a (IC50 40 nM) is 5,000 times higher than that of the prohormone of calcitonin (Km,app 200 μM). Whatever the difference between the DMS53 cellular enzyme and that secreted into the medium, it markedly affects the efficiency of inhibition by the glycolate 1a but causes little change to the binding affinities of either procalcitonin or N-decanoylglycine 1c. As a result, the glycolate 1a is not only a potent inhibitor of the mammalian cellular protein, it is also selective for inhibition of the cellular enzyme, to which it binds around three orders of magnitude more strongly than either the analogous substrate 1c or procalcitonin.
Acknowledgements
We gratefully acknowledge financial support received from the Australian Research Council, and technical assistance of Cheng Sun with compound synthesis and characterisation, and support and advice from Dr Mario Lobigs regarding cell line culture.Notes and references
- D. J. Merkler, Enzyme Microb. Technol., 1994, 16, 450–456 Search PubMed.
- S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 1997, 278, 1300–1305 CrossRef CAS.
- S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Nat. Struct. Biol., 1999, 6, 976–983 CrossRef CAS.
- S. T. Prigge, R. E. Mains, B. A. Eipper and L. M. Amzel, Cell. Mol. Life Sci., 2000, 57, 1236–1259 CAS.
- S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864–867 CrossRef CAS.
- D. A. Groneberg, J. Springer and A. Fischer, Pulm. Pharmacol. Ther., 2001, 14, 391–401 Search PubMed.
- Z. Wiesenfeld-Hallin and X.-J. Xu, Eur. J. Pharmacol., 2001, 429, 49–59 Search PubMed.
- (a) A. V. Schally, A. M. Comaru-Schally, A. Nagy, M. Kovacs, K. Szepeshazi, A. Plonowski, J. L. Varga and G. Halmos, Front. Neuroendocrinol., 2001, 22, 248–291 Search PubMed; (b) J. Du, B. P. Keegan and W. G. North, Cancer Lett., 2001, 165, 211–218 Search PubMed; (c) N. Jiménez, J. Jongsma, A. Calvo, T. H. van der Kwast, A. M. Treston, F. Cuttitta, F. H. Schröder, L. M. Montuenga and G. J. van Steenbrugge, Int. J. Cancer, 2001, 94, 28–34 Search PubMed.
- (a) B. J. Wilcox, K. J. Ritenour-Rodgers, A. S. Asser, L. E. Baumgart, M. A. Baumgart, D. L. Boger, J. L. DeBlassio, M. A. deLong, U. Glufke, M. E. Henz, L. King III, K. A. Merkler, J. E. Patterson, J. J. Robleski, J. C. Vederas and D. J. Merkler, Biochemistry, 1999, 38, 3235–3245 CrossRef CAS; (b) C. R. Hiley and P. M. Hoi, Cardiovasc. Drug Rev., 2007, 25, 46–60 Search PubMed; (c) G. P. Mueller and W. J. Driscoll, Vitam. Horm., 2009, 81, 55–78 Search PubMed.
- A. F. Bradbury, J. Mistry, B. A. Roos and D. G. Smyth, Eur. J. Biochem., 1990, 189, 363–368 CAS.
- A. G. Katopodis and S. W. May, Biochemistry, 1990, 29, 4541–4548 CrossRef CAS.
- F. N. Bolkenius, A. J. Ganzhorn, M.-C. Chanal and C. Danzin, Biochem. Pharmacol., 1997, 53, 1695–1702 Search PubMed.
- J. A. Trendel, N. Ellis, J. G. Sarver, W. A. Klis, M. Dhananjeyan, C. A. Bykowski, M. D. Reese and P. W. Erhardt, J. Biomol. Screening, 2008, 13, 804–809 Search PubMed.
- G. P. Mueller, W. J. Driscoll and B. A. Eipper, J. Pharmacol. Exp. Ther., 1999, 290, 1331–1336 Search PubMed.
- A. A. Ogonowski, S. W. May, A. B. Moore, L. T. Barrett, C. L. O'Bryant and S. H. Pollock, J. Pharmacol. Exp. Ther., 1997, 280, 846–853 Search PubMed.
- J. D. Bauer, J. A. Sunman, M. S. Foster, J. R. Thompson, A. A. Ogonowski, S. J. Cutler, S. W. May and S. H. Pollock, J. Pharmacol. Exp. Ther., 2007, 320, 1171–1177 Search PubMed.
- E. Langella, S. Pierre, W. Ghattas, M. Giorgi, M. Réglier, M. Saviano, L. Esposito and R. Hardré, ChemMedChem, 2010, 5, 1568–1576 Search PubMed.
- M. D. Andrews, K. A. O'Callaghan and J. C. Vederas, Tetrahedron, 1997, 53, 8295–8306 Search PubMed.
- J. Feng, J. Shi, S. R. Sirimanne, C. E. Mounier-Lee and S. W. May, Biochem. J., 2000, 350, 521–530 Search PubMed.
- (a) L. A. Miller, L. E. Baumgart, G. H. Chew, M. A. deLong, L. C. Galloway, K. W. Jung, K. A. Merkler, A. S. Nagle, D. D. Poore, C. H. Yoon and D. J. Merkler, Arch. Biochem. Biophys., 2003, 412, 3–12 Search PubMed; (b) N. R. McIntyre, E. W. Lowe Jr., G. H. Chew, T. C. Owen and D. J. Merkler, FEBS Lett., 2006, 580, 521–532 Search PubMed.
- D. J. Merkler, A. S. Asser, L. E. Baumgart, N. Carballo, S. E. Carpenter, G. H. Chew, C. C. Cosner, J. Dusi, L. C. Galloway, A. B. Lowe, E. W. Lowe Jr., L. King III, R. D. Kendig, P. C. Kline, R. Malka, K. A. Merkler, N. R. McIntyre, M. Romero, B. J. Wilcox and T. C. Owen, Bioorg. Med. Chem., 2008, 16, 10061–10074 Search PubMed.
- D. Schade, J. Kotthaus, H. Hungeling, J. Kotthaus and B. Clement, ChemMedChem, 2009, 4, 1595–1599 CrossRef CAS.
- M. D. Erion, J. Tan, M. Wong and A. Y. Jeng, J. Med. Chem., 1994, 37, 4430–4437 CrossRef CAS.
- D. Ping, C. E. Mounier and S. W. May, J. Biol. Chem., 1995, 270, 29250–29255 Search PubMed.
- B. J. W. Barratt, C. J. Easton, D. J. Henry, I. H. W. Li, L. Radom and J. S. Simpson, J. Am. Chem. Soc., 2004, 126, 13306–13311 Search PubMed.
- Although KI, IC50 and KM, app values are different parameters, they are suitable for comparison as approximate measures of enzyme binding affinity, as used in the present work.
- (a) J. A. Trendel, N. Ellis, J. G. Sarver, W. A. Klis, M. Dhananjeyan, C. A. Bykowski, M. D. Reese and P. W. Erhardt, J. Biomol. Screening, 2008, 13, 804–809 Search PubMed; (b) M. D. Vos, J. E. Jones and A. M. Treston, Gene, 1995, 163, 307–311 CrossRef CAS; (c) L'H. Ouafik, S. Sauze, F. Boudouresque, O. Chinot, C. Delfino, F. Fina, V. Vuaroqueaux, C. Dussert, J. Palmari, H. Dufour, F. Grisoli, P. Casellas, N. Brünner and P.-M. Martin, Am. J. Pathol., 2002, 160, 1279–1292 Search PubMed.
- (a) S. L. Milgram, R. C. Johnson and R. E. Mains, J. Cell Biol., 1992, 117, 717–728 Search PubMed; (b) C. Rajagopal, K. L. Stone, R. E. Mains and B. A. Eipper, J. Biol. Chem., 2010, 285, 34632–34642 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of synthesis and characterisation of compounds 2a, 3a, 5a–d, 1b–4b, 6a–b and 1c–3c, preparation of PAM isolates and assays for enzyme activity. See DOI: 10.1039/c1md00079a |
| ‡ On placement from the Department of Chemistry, University of Aberdeen. |
| This journal is © The Royal Society of Chemistry 2011 |