Towards kit-like 18F-labeling of marimastat, a noncovalent inhibitor drug for in vivo PET imaging cancer associated matrix metalloproteases†
Ying
Li
a,
Richard
Ting
a,
Curtis W.
Harwig
a,
Ulrich
auf dem Keller
b,
Caroline L.
Bellac
b,
Philipp F.
Lange
b,
James A. H.
Inkster
c,
Paul
Schaffer
c,
Michael J.
Adam
ac,
Thomas J.
Ruth
c,
Christopher M.
Overall
b and
David M.
Perrin
*a
aDepartment of Chemistry, The University of British Columbia, 2036 Main Mall. Vancouver, BC, Canada. E-mail: dperrin@chem.ubc.ca; Fax: +1-604-822-2847; Tel: +1-604-822-0567
bCentre for Blood Research and Department of Biochemistry and Molecular Biology, The University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC, Canada. E-mail: chris.overall@ubc.ca; Fax: +1-604-822-7742; Tel: +1-604-822-2958
cTRIUMF, 4004 Wesbrook Mall, Vancouver, BC, Canada. E-mail: truth@triumf.ca; Tel: +1-604-222-7526
First published on 7th July 2011
Abstract
Marimastat, a clinically trialed drug developed to treat breast cancer by inhibiting cancer-associated matrix metalloproteases (MMPs), was linked to an aryl boronic ester for single-step [18F]-aqueous fluoride capture and the labeled product revealed tumor associated MMP activity in vivo. Herein, we report important radiosynthetic attributes for labeling marimastat that enabled the first PET images of breast cancer-associated matrix metalloproteases in a syngenic murine model. The advantages of this method include one-step post synthetic labeling in less than one hour at ambient temperature, the ability to work in aqueous media without drying the 18F-fluoride, observation of high radiochemical purity, and the potential for tripling the specific activity of the fluoride used in labeling. Using low levels of activity e.g. 60 mCi in low volumes this method affords reasonable yields of labeled marimastat with decay-corrected specific activities of 0.39 and 0.75 Ci/μmol, and real specific activities of 0.16 and 0.39 Ci/μmol. Current limitations of this method along with anticipated improvements are discussed.
Introduction
Our understanding of diseases at the molecular level is being driven by powerful techniques in the field of molecular imaging that allow non-invasive determination of functional changes of a given physiological state, often long before gross anatomical changes are evident. By quantifying changes at the molecular level, health researchers can provide more accurate and earlier diagnoses as well as gauge the efficacy of individual patient treatment. The confluence of new proteomic/genomic probes along with large combinatorial libraries has revealed many new cellular markers and provided vast numbers of potential targeting agents, which when suitably radiolabeled, will enable the visualization of deep biological effects obscured by tissue. To date, numerous imaging probes, of which a growing number involve the use of peptide-based ligands and other large molecules, have been developed and studied using SPECT (single photon emission computed tomography) and PET (positron emission tomography).While convenient, rapid, and readily reproducible radiochemical labeling methods make SPECT an effective means of imaging peptides, the increasingly important application of PET imaging is predicated on similar conditions: a) a suitable ligand that binds with high specificity and affinity to a cellular biomarker that is present at increased levels in pathologically altered cells or tissue, and b) radiosynthetic methods to label the ligand with a suitable positron-emitting nuclide so that PET can be used to map distribution, target localization, and monitor in vivo clearance.1,2 Among the various positron emitting radionuclides such as 64Cu, 68Ga, 76Br, 89Zr, 94mTc, and 124I that are being explored for PET imaging, the several favorable nuclear attributes of 18F, including high isotopic purity, a single decay process, low β+ emission energy, and facile production account for its widespread use in the field.
Nevertheless, the relatively short half-life of 18F (109.8 min) necessitates expedient labeling methods, which in turn pose several challenges for biomolecular labeling, particularly due to the high solvation energy of fluoride, which limits its nucleophilicity in water wherein most mid-size and larger biomolecules must be stored. Generally, [18F]-fluoride is punctiliously dried to reveal its nucleophilicity, whereupon it is reacted at high temperature (80–140 degrees) in polar-aprotic solventse.g.DMSO.3 Whereas such conditions are suitable for direct labeling of small nonpolar organic ligands,4,5 these conditions are usually incompatible with labeling larger, water soluble peptides and related biomolecules, which typically require at least one additional bioconjugation step involving an 18F-labeled prosthetic precursor.
An increasing number of reports have focused on aqueous and/or single-step incorporation of fluoride into biomolecular constructs. Direct C–F,6,7 Al–F,8 Si–F,9–11 and B–F12 bond forming strategies have all received considerable attention.13,14 Towards these ends, we previously described a biotinylated conjugate of 2,4,6-tri-fluoro-3-(4,4,5,5-tetraphenyl-1,3,2-dioxaborolan-2-yl)benzamide as a precursor for aqueous 18F-capture that provided a biotinylated aryltrifluoroborate (ArBF3−) salt with a specific activity of <0.1 Ci/μmol that was stable in vivo.15 Due to their high water solubility, high polarity/low lipophilicity, and rapid clearance, 18F-labeled ArBF3− conjugates represent potentially useful 18F-PET imaging agents.16
Labeling occurs according to eqn (1): three fluoride ions condense with one boronic acid/ester (R = alkyl or H) under acidic conditions to afford an ArBF3−.
| ArB(OR)2 + K[18/19F] + 2H[18/19F] → ArB[18F][19F]2K + 2ROH | (Eq. 1) |
Acid must be added as a catalyst to protonate the OR groups, which cannot leave as alkoxide or hydroxide. The addition of acid also converts KF to KHF2 and HFin situ. The decay-corrected specific activity of the ArBF3− is triple that of the [18F]-fluoride, which is a direct consequence of the stoichiometry of eqn (1) in accordance with the law of conservation of mass. Indeed, because specific activity of the ArBF3 is defined as Ci/micromole ArBF3, then eqn (2) states:
| (Ci/μmol ArBF3) = (Ci/μmol F) × (3 μmol F/μmol B) | (Eq. 2) |
A graphical representation of this is found in the ESI.† This holds true for alllabeling regimes, including no-carrier-added preparations, which always contain substantial amounts of carrier [19F]-fluoride (vide infra). Stochastically however, there is only one [18F]-fluoride atom per ArBF3− (discussed extensively in ref. 13, 24 and 25).
When relatively low activity is used e.g. ∼60 mCi, both reasonable yields and useful specific activities are achieved in low volumes that minimize the addition of carrier [19F]-fluoride required to maintain a relatively high fluoride concentration. Although working in low volumes might appear challenging to some, herein we show that this method can be used to label medically important peptidic ligands in a kit-like manner that preserves bioactivity while affording radiochemically pure material in reasonable yields with specific activities suitable for imaging.
To demonstrate the broader applicability of this method for labeling a peptidic drug (as opposed to biotin), we labelled marimastat, an orally bioavailable, clinically trialed non-covalent nanomolar broad-spectrum inhibitor of zinc-dependent matrix metalloproteinases (MMPs) that passed preliminary toxicity trials yet failed in late stage because of lack of efficacy and unwanted side effects. The development of marimastat was guided by the finding that elevated MMP activity is pathognomonic of inflammatory disease and in particular, cancer; MMPs that normally control extracellular homeostasis are activated to remodel the extracellular matrix, causing tissue destruction and enhancing metastasis and carcinogenesis.17–19 Based on marimastat's rich clinical history, we selected it (Fig. 1) to provide some of the first images of tumor associated MMPs in mice,20 of which there are few examples, and thereby create a potentially new imaging agent from a trialed drug candidate.21–25 While these PET images have been published, the important aspects of the radiosynthetic methods are presented here.
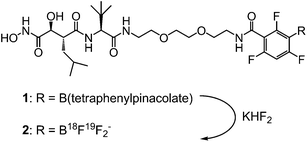 | ||
| Fig. 1 Marimastat-ArB(OR)2 (1) and marimastat-ArBF3K (2). | ||
Results
Synthesis and purification
Marimastat was conjugated via a glycol linker to an arylboronyltetraphenylpinacolate15,26 as noted in Scheme 1 (ESI†), to afford 1, which following HPLC purification, was lyophilized, stored in 100 nmol aliquots (98 μg), and used as needed.[18F]-fluoride was prepared as noted in the methods section. Typically, a total of 950 nmols of [19F]-fluoride along with various chemoprotectants (ascorbate, formate, and iodide) were added to ∼300 mCi no-carrier-added [18F]-fluoride to give a fluoride cocktail of 4.5 μLwith a defined specific activity of 0.251 Ci/μmol at the start of synthesis (it is assumed that the NCA [18F]-fluoride had negligible amounts of carrier). From this cocktail, 1.5 μL containing ∼60 mCi (251 nmol) was added to 100 nmol 1 contained in 1.5 μL MeCN, bringing the final concentration of fluoride to 72 mM.
Addition of concentrated HCl effectively neutralized the co-eluted carbonate to bring the pH to 1–2, which is partially buffered by a KHF2/HF buffer system. Acid catalyzes ArBF3− formation, which proceeded for 45 min at r.t (17–19 °C) in aqueous MeCN (∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). The reaction was quenched by addition of aqueous NH3 to bring the pH to ∼7 and the mixture (∼100 μL) containing ∼40 mCi was injected onto an analytical RPC18 HPLC (Fig. 2). 18F-labeled 2 (1.1 mCi, 2 mL) was separated from the much less polar precursor 1 that elutes much later (data not shown). Labeling proceeded with exceptional chemoselectivity to afford a single radiolabeled peak with no observed radiolytic products. The pooled fractions containing 18F-labeled 2 were concentrated by rotary evaporation with mild heating 45–50 °C to provide just under 1 mCi for animal imaging after a total operation time of 105 min.
:50). The reaction was quenched by addition of aqueous NH3 to bring the pH to ∼7 and the mixture (∼100 μL) containing ∼40 mCi was injected onto an analytical RPC18 HPLC (Fig. 2). 18F-labeled 2 (1.1 mCi, 2 mL) was separated from the much less polar precursor 1 that elutes much later (data not shown). Labeling proceeded with exceptional chemoselectivity to afford a single radiolabeled peak with no observed radiolytic products. The pooled fractions containing 18F-labeled 2 were concentrated by rotary evaporation with mild heating 45–50 °C to provide just under 1 mCi for animal imaging after a total operation time of 105 min.
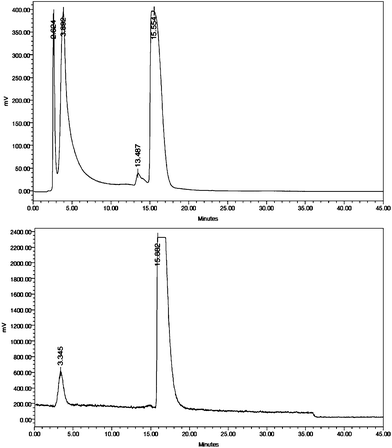 | ||
| Fig. 2 A) HPLC radiochromatogram of the crude reaction (low sensitivity mode), 18F-labeled 2 elutes starting at 15.5 min; B) analytical HPLC radiochromatogram of sample containing fraction 35 and 2.5% of pooled fractions (high sensitivity mode). | ||
Yield, purity, and relationship to image quality
Although the HPLC traces suggest radiochemical yields in excess of 50%, the isolated radiochemical yield of 18F-labeled 2 was ∼2.75%, while the yield in terms of conversion of 1 to 2 was ∼2.6%. A second synthesis produced 18F-labeled 2 in similar yield and radiochemical purity, with a specific activity of 0.19 Ci/μmol (see the ESI†).Although the chromatographic yield should equal the isolated yield, the former usually surpasses the latter. Two explanations delimit the discrepancy between the chromatographic and isolated yields seen herein: i) large amounts of unincorporated fluoride stuck to the column and never eluted through the detector giving the appearance of a 50% yield when in fact the lower isolated yield reflects the true yield, or ii) both[18F]-fluorideand18F-labeled 2 stuck to the column such that the chromatographic yield reflects a yield that should be realized by using alternative purification approaches. Indeed, by TLC we have observed higher yields (10–30%) in terms of the conversion of arylboronate esters to ArBF3−s under these conditions, which suggests that nonspecific retention of 18F-labeled 2 explains the lower isolated yield.
Whereas the purification will require further optimization to improve yields, the chemoselectivity of this method affords a single radiolabeled species that facilitates chromatographic peak picking. In addition, while this method proceeds in the presence of various chemoprotectants (ascorbate, iodide, formate), which can be added to prevent radiolysis, in subsequent labelings we found that they were not needed (data not shown). Finally, we circumvent fluoride desiccation that precedes the synthesis of an 18F-labeled precursor that is conjugated to the biomolecule in at least one additional step followed by HPLC purification.
Other methods produce similar products in similar yields via more involved procedures. For instance, starting with 800 mCi, NCA [18F]-fluoride, a peptide nucleic acid was labeled in multistep fashion in approximately 10% isolated yield after 80–90 min.27 Similarly, an oligonucleotide was labeled in two steps with an overall isolated yield of ∼4% after a total time of 276 min.28 Finally, a recent one-step labeling of RGD at 130 °C resulted in several radiolabeled species from which the desired compound was purified by HPLC in yields ranging from 9–27%.7
Another notable difference between this method and more standard approaches is that standard approaches require up to 3 mg of precursor ligand7,11 in order to ensure relatively rapid labeling (pseudo-first order kinetics) in standard volumes (0.1–1 mL). Herein, labeling proceeds with less than 100 μg of precursor. In the case of peptide-[19F]-silylfluoride bioconjugates, which have been elegantly designed to undergo 18F-19F isotope exchange, [19F]-fluoride containing precursor, as carrier, is present at the outset of radiosynthesis, although impressively low quantities of carrier ligand are used to ensure relatively high specific activity. As in almost all cases, HPLC purification is required, irrespective of the labeling chemistry.
In terms of purity, a small (∼5%) contamination of free [18F]-fluoride is observed in the analytical trace (Fig. 2B) following purification, and is likely due to imperfect separation from free fluoride or slow solvolysis of the labeled compound that occurred upon standing for ∼2 h prior to analytical HPLC analysis, and which might have been further accelerated by heating the sample to ∼50 degrees during just rotary evaporation prior to packaging. 19F-NMR spectroscopy allowed us to establish in this case the rate for solvolytic fluoride loss from 19F-labeled 2 is very slow (ksolv. = 5.61 × 10−4 min−1, t1/2 ∼ 1230 min, see the ESI†). This rate constant is nearly identical to constants we measured previously for other similarly linked ArBF3−s.15,29
Animal images of tumor-associated MMP activity were acquired with 18F-labeled 2, the synthesis of which is reported here.20 In order to visualize the tumors, the contrast was greatly enhanced such that some bone uptake was observed. Bone localization was attributed to minor amounts of free [18F]-fluoride, as seen in the analytic traces of Fig. 2, or to uptake of 18F-labeled 2 itself. Notably, no improvement in standard uptake values or signal-to-background ratios was seen with three-fold increased specific activity suggesting that the inability to obtain high contrast tumor images may have more to do with marimastat's lack of specificity for tumor associated proteases compared to serum targets, than with the 18F-labeled ArBF3− appendage.20
Specific activity
Since the specific activity of the [18F]-fluoride was set at 0.25 Ci/μmol, eqns (1) and (2) provide a decay-corrected specific activity of 0.75 Ci/μmol for 18F-labeled 2. Following a total time 105 min (start-of-synthesis to end of packaging) the specific activity of 18F-labeled 2 decays to 0.39 Ci/μmol. Radioisotopic dilution of this sort provides the most accurate method for concentration determination. Thus, the final concentration of 18F-labeled 2 is calculated from its specific activity at end of packaging, divided by the sample volume. Based on this method, 18F-labeled 2 exhibits an apparent IC50 of ∼2 nM against MMP2 activity. These findings are consistent with previous reports that marimastat derivatives, appended through the carboxy terminus, exhibit low nanomolar IC50 values against MMP2, which are comparable to marimastat itself.30To independently verify the IC50 values, we first calculated the concentrations of various marimastat derivatives using a colorimetric ferroin assay31–33 and thereby measured the IC50 values of 19F-labeled 2, as well as a Cbz-protected glycol-linked marimastat precursor (compound 12, Scheme 1, ESI†), which provided an additional control to demonstrate the ineffectual nature of the linker arm on the IC50 value. 19F-labeled 2, compound 12, and marimastat respectively exhibit IC50 values of 2.19 nM, 1.88 nM, and 2.05 nM using a standard MMP2 assay.20
Beyond purity and yield, “effective specific activity”,34 defined as Ci per micromole of labeled ligand, is the most relevant criterion for imaging. While the specific activities reported herein are modest, the potential for procuring 18F-labeled ArBF3− bioconjugates with high specific activity, along with the imperfect relationship between specific activity and image quality merits the following discussion. Whereas carrier-free [18F]-fluoride has a specific activity of 1712 Ci/μmol, in practice, the specific activity of “no-carrier-added” (NCA) [18F]-fluoride commonly falls in the range of 3–10 Ci/μmol. This value places a limit on attainable specific activities and confirms the presence of significant amounts of carrier [19F]-fluoride in all[18F]-fluoride preparations.34–36 The decay that accompanies multistep radiosyntheses results in a further erosion of the final specific activities of most radiopharmaceuticals, which commonly fall below 2.0 Ci/μmol.37
While these practical limits befall all radiosyntheses, one must question the existence of an “ideal” specific activity for PET imaging, particularly because PET images of tumors in humans38–40 and animals41,42 are obtained using ligands with specific activities of 0.017 Ci/μmol,43 0.08 Ci/μmol,44 and 0.25 Ci/μmol.45,46 While these values are clearly suitable for clinical imaging they fall far short of practical limits, not to mention falling exceedingly short of the theoretical limit. Thus, while there appears to be no corroboration of an “ideal” specific activity, a specific activity of ∼0.5 Ci/μmol is likely to be largely sufficient for imaging purposes47 and thus probably represents a benchmark minimum that any labeling technique should ultimately meet. Gratifyingly, our method attains this value, despite the use of relatively low levels of activity e.g. 63 mCi.
The relationship between specific activity and image quality is further complicated by specific and nonspecific binding to nontarget humeral proteins;48 that is to say, in some cases, the presence of unlabeled ligand (carrier) can be tolerated, as in the case of 18F-labeled insulin.49 In other cases, the addition of carrier ligand actually enhances image quality, as in the case of images of ligand uptake in somatostatin receptor-positive organs using carrier-doped 111In-labeled penetreotide.50 Thus, a dogmatic insistence on no-carrier-added labelings may be unjustified.
Nevertheless, for those who would insist on very high specific activities, we discuss how this method can provide 18F-labled ArBF3−s with specific activities that approach or even surpass the aforementioned practical limits of 2 Ci/μmol. Eqn (1) implies scalability: starting with multi-Curie levels of [18F]-fluoride common to production labs,51,52 a labeling reaction with 600 mCi of [18F]-fluoride, where the specific activity is deliberately set at 2 Ci/μmol, will contain 300 nmol of total fluoride, which in 4 μL, represents 75 mM total fluoride, a concentration slightly higher than that used herein. Under such conditions, the resulting ArBF3− will be produced with a decay-corrected specific activity of 6 Ci/μmol (∼3 Ci/μmol at packaging time) in yields of at least 15 mCi. Increasing the volume to a more practical 10 μL while maintaining the fluoride concentration at 72 mM will reduce the decay-corrected specific activity of the ArBF3− to 2.5 Ci/μmol while leaving yields unchanged. Alternatively, with a decay-corrected specific activity set for 2.5 Ci/μmol and a volume set at 4.0 μL, the fluoride concentration can be increased to 180 mM, which will greatly increase yields. A recent report on microreactor technology demonstrated 99.5% elution of 876 mCi [18F]-fluoride (10 Ci/μmole) from a 2 μL anion exchange column into a volume of 5 μL.53 This report dispels any doubts as to possibility of manipulating large quantities of [18F]-fluoride in low volumes. With no need to dry the fluoride prior to synthesis, the production of ArBF3−s at high specific activity is readily contemplated.
While to date small volumes for radiolabeling may not be widely used, small volumes are commonly used in manual and automated kits for PCR and DNA sequencing. Without doubt, these will find increasing use for PET chemistry. Indeed, the application of nanoreactors, which have recently manipulated nanolitre volumes of [18F]-fluoride,53–55 may provide even higher specific activities and yields that rely solely on the carrier [19F]-fluoride inherently present in NCA [18F]-fluoride preparations. Provided that 50 mCi [18F]-fluoride at 10 Ci/μmol can be captured in 50 nL, the fluoride concentration would be 100 mM, which will afford an 18F-labeled ArBF3− in greater yields that those herein and with a decay-corrected specific activity of 30 Ci/μmol. Indeed, the use of nanoreactors will enhance ArBF3− yields. More interesting yet, because of the tripling of specific activity, nanoreactors will enable the synthesis of ArBF3−conjugates with specific activities that simply cannot be achieved with standard methods.
Conclusions
In summary, this method embodies several advantages for 18F-labeling peptide tracers. These include: 1) single step kit-like labeling; 2) use of aqueousfluoride that need not be scrupulously dried, but only concentrated in water or mixed aqueous co-solvents; 3) the ability to work at room temperature; 4) yields on par with standard labeling methods and specific activities within the range of clinical use; 5) the need for only microgram amounts of a shelf-stable precursor to produce suitable quantities of a radiochemically pure 18F-labeled ArBF3− conjugate (>95%); 6) a tripling of the specific activity, decay-corrected to start of synthesis, which may prove very advantageous when using production level quantities of [18F]-fluoride.In addition, this one-step method now avoids some of the other drawbacks associated with multi-step syntheses, namely the requirement to bring [18F]-fluoride to dryness, to work at highly elevated temperature, and to isolate radiolabeled prosthetics for subsequent bioconjugation. Although current reaction times, yields, and specific activities are slightly lower than traditional multistep labeling approaches, these aspects are likely to improve with slight modifications to purification methods that avoid HPLC in favor or cartridge based approaches. Further optimization of yields through chemical modifications to the leaving OR-groups that depart more readily than the tetraphenylpinacol, and to the aryl nucleus to increase the rate of fluorination and reduce the rate of solvolysis56 respectively are being explored. Finally, this highly chemoselective one-step aqueous 18F-labeling method should be generally applicable for labeling peptides, peptoids, and even larger molecules including affibodies and affilins that are exceptionally stable at pH 2 or lower.57 One-pot, two-step labeling reactions involving click chemistry are being explored for molecules that may not resist low pH. These improvements will be reported in the near future.
Materials and methods
General
Chemicals were purchased from Sigma-Aldrich and Acros Organics. Deuterated solvents were purchased from Cambridge Isotope Laboratories. Analytical and preparative thin layer chromatography was performed using Silica Gel 60 F254 Glass TLC plates from EMD Chemicals. All HPLC solvents were filtered prior to use. Compound 1 was purified by HPLC using an Agilent 1100 HPLC system equipped with a diode array UV detector. Column used was either a semi-preparative Agilent Eclipse XDB C18 5μm 9.4 × 250mm, or an analytical Phenomenex Jupiter 10μ C18 300 Å 4.60 × 250mm, as noted in specific sections. All reverse phase radio-HPLC reported herein utilized a Waters 600 Controller in combination with a Waters 2487 Dual λ Absorbance Detector and a Bioscan NaI detector and an analytical Agilent Eclipse XDB C18, 5μm 4.6 × 150mm. HPLC purification system 1: solvent A: H2O, solvent B: MeOH, step gradients: 0% B from 0 to 2 min., then linear gradient from 0% to 10% B from 2 to 3 min., 10% to 50% B from 3 to 10 min., 50% to 100% B from 10 to 20min., 100% B from 20 to 40 min., 100% to 0% B from 40 to 45 min; flow rate: 1 mL min−1; room temperature 17–19 °C. HPLC system 2: solvent A: 0.1% TFA in H2O, solvent B: 0.05% TFA in MeCN, step gradients: 10% B from 0 to 2 min., 10% to 50% B from 2 to 20 min., 50% to 100% B from 20 to 25 min., 100% B from 25 to 28 min., 100% to 10% B from 28 to 30 min., 10% B from 30 to 32 min; flow rate: 1 mL min−1; 50 °C. HPLC system 3: solvent A: 0.1% TFA in H2O, solvent B 0.05% TFA in MeCN step gradients: 10% B from 0 to 2 min., 10% to 50% B from 2 to 5 min., 50% to 100% B from 5 to 8 min., 100% B from 8 to 12 min., 100% to 10% B from 12 to 15 min., 10% from B 15 to 16 min.; flow rate: 3 mL min−1; 50 °C. All 1H-NMR and 19F-NMR spectra were recorded on a Bruker Avance 300 or 400 MHz instrument. Chemical shifts are reported using the δ scale in ppm and all coupling constants (J) are reported in hertz (Hz). Unless specified, 1H NMR spectra are referenced to the tetramethylsilane peak (δ = 0.00) and 19F-NMR spectra are referenced to NEAT trifluoroacetic acid (δ = 0.00, −78.3 ppm relative to CFCl3). Due to the presence of 19F-fluoride contamination in the NMR spectrometer probe, baseline corrections for samples less than 20 mM in [19F]-fluoride concentration had to be adjusted by multipoint linear baseline correction using MestReC 4.9.9.9 or MestReNova 6.0. This correction did not affect the absolute chemical shifts or integration ratios of 19F signals. All HRMS readings were acquired on a MicromassLCT.Radiochemistry
[18F]-fluoride was prepared as noted in the methods section. Typically, at end of bombardment, ∼300 mCi no-carrier-added (NCA) was obtained. To this NCA [18F]-fluoride was added 700 nmol [19F]-fluoride (KHF2 form) contained in 1 μL and then concentrated to a visibly moist residue with a helium purge (∼10 min, 110 °C) and resuspended in a solution (4.5 μL) containing an additional 250 nmol [19F]-fluoride (KHF2 form), along with 1 M ascorbic acid, 0.2 M KI, and 0.2 M ammonium formate, which were present to prevent adventitious radiolysis during labeling. Typically, the pH of this solution is 9–11 and reflects the gross excess of CO32− that coelutes with the [18F]-fluoride. This gave a fluoride cocktail with a defined specific activity of 0.251 Ci/μmol at the start of synthesis. From this cocktail, 1.5 μL containing ∼63 mCi (251 nmol) was added to 100 nmol 1 contained in 1.5 μL MeCN.Addition of concentrated HCl (0.5 μL) brought the pH to 1–2 (as verified by spotting 0.1μL on pH paper) and the reaction was allowed to proceed cleanly for 45 min at rt (∼18 °C). The reaction was then quenched by the addition of 0.3% aqueous NH3 (105 μL) to bring the pH to ∼7. Approximately 100 μL of the quenched crude reaction mixture containing ∼40 mCi was purified by direct injection onto an analytical RPC18 HPLC (Fig. 2) using a 45 min. step gradient elution of 0%–100% H2O-MeOH at 1 mL/min. with fractionation each 0.5 min. 18F-labeled 2 (1.1 mCi, 2 mL) was separated from the much less polar precursor 1 that eluted much later (data not shown). The pooled fractions containing 18F-labeled 2 were concentrated by rotary evaporation with mild heating 45–50 °C to provide just under 1 mCi for animal imaging after a total of 105 min from the initiation of synthesis until packaging.
A separate labeling that provided 18F-labeled 2 at lower specific activity, was similarly performed. 212 mCi of NCA [18F]-fluoride was concentrated to eye-apparent dryness in a 5 mL Pyrex V-vial at 110 °C under helium purge. After dry down, 4 μL of water was added into the V-vial and 2 μL (103 mCi) of the resuspended NCA [18F]-fluoride was removed for other purposes unrelated to this work. To the remaining 109 mCi NCA [18F]-fluoride was added 800 nmols of [19F]-fluoride (KHF2 form) contained in 42 μL of water (0.109 Ci/0.8μmol = 0.136 Ci/μmol). The resulting solution was then reconcentrated to eye-apparent dryness under helium flow at which point there was 109 mCi (4 min. prior to the start of synthesis) in the V-vial. Following this re-concentration, a solution (3 μL) of 1 M ascorbic acid, 0.2 M potassium iodide, and 0.2 M ammonium formate was added to the V-vial. A volume of this fluoride cocktail (2 μL) was transferred to a PCR tube containing 100 nmols 1 in 2 μL acetonitrile (97 mM total fluoride) and this reaction was found to contain 53 mCi. Labeling was initiated by the addition of acid as previously noted. After 87 min. at room temperature (17–19 °C) for the reaction was quenched as noted above and loaded directly in an analytical reverse phase HPLC analytical column that afforded 1.1 mCi. of 18F-labeled 2 at a final specific activity of 0.185 Ci/μmol corrected for 126 min time between the start of synthesis and the end of packaging.
This work, as conducted and reported herein, involved the potential for partial and whole-body exposure to β+ and γ-ray irradiation. While we have safely used low level amounts of radiation (e.g. 5–10 mCi) to reproduce the results below numerous times with low specific activities, the two radiosyntheses, which afforded imaging-quality, decay-uncorrected specific activities of 0.19 Ci/μmol or 0.39 Ci/μmol, involved a manual transfer of 30–60 mCi from even higher quantities (200 mCi). While high milliCurie levels of [18F]-fluoride, and indeed even multi-Curie levels as reported elsewhere, are commonly used in automated production labs that have suitable infrastructure shielding, current shielding limitations at the Triumf-PET production facility prevented us from obtaining high milliCurie levels of radiation for this work. The radiosyntheses described herein were performed manually by hand-pipetting in a hot cell that was too small to contain an HPLC system. Despite security measures that included wearing lab coats, safety goggles, double sleeves, boots, and triple gloves, whole body dosimeters worn during this work registered minimal yet detectable whole body exposure.
Chemical syntheses
Compounds 3–6 (synthetic scheme—ESI†) were prepared according to literature precedent. Briefly, Boc-L-tert-leucine30 was coupled to the mono-Cbz-protected diamine 358 using TBTU to give 4 in high yield. Removal of the Boc-protecting group with TFA gave the desired amine salt, which was used immediately in the subsequent coupling step. The acetonide-protected diacid 6 was synthesized from (2S,3R)-2-hydroxy-3-isobutylsuccinic acid30 by treatment with catalytic p-toluene sulfonic acid (PTSA) in 2,2-dimethoxypropane.59 Coupling of 5 and 6 under epimerization-free conditions60 afforded 7 in 74% yield and conversion of the acetonide 7 to methyl ester 8 was achieved by transesterification in the presence of catalytic PTSA in refluxing MeOH and subsequently hydrogenolysis to remove the Cbz-protecting group to give 9. Final attachment of the resultant amine 9 to 2,4,6-trifluoro-3-carboxyphenylboronic tetraphenylpinacolyl ester 1015,26 by EDC coupling gave the methyl ester precursor 11. Treatment of 11 with hydroxylamine by the method of Hoet al.61 gave the desired hydroxamate title precursor compound 1 for18F-labeling. Partial loss of the tetraphenyl-1,3,2-dioxaborolan-2-yl group by deboronation during treatment of 11 with aqueous hydroxylamine accounted for the low yields (15–30%) obtained for 1.:1 then 2:1 then 1:0) to afford the desired adduct 4 (2.24 g, 94%) as a clear oil. 1H NMR (300 MHz, CDCl3) δ 7.34–7.29 (m, 5H), 6.24 (br s, 1H), 5.51 (br s, 1H), 5.31 (m, 1H), 5.10 (s, 2H), 3.79 (br d, J = 7.3 Hz, 1H), 3.59–3.40 (m, 12H), 1.41 (s, 9H), 0.96 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 170.7, 156.4, 155.6, 136.4, 128.2, 127.9, 127.8, 79.2, 70.0, 69.8, 69.4, 66.4, 62.0, 40.6, 38.8, 34.3, 28.1, 26.3; HRMS (ESI) calcd. for C25H41N3O7Na+ (M + Na)+ 518.2842, found 518.2843.
:2 then 96:4) to afford the desired adduct 7 (134 mg, 74%) as a pale yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.33–7.28 (m, 5H), 6.63 (m, 1H), 6.31 (m, 1H), 5.51 (m, 1H), 5.10 (s, 2H), 4.44 (d, J = 6.2 Hz, 1H), 4.17 (br d, J = 9.2 Hz, 1H), 3.59–3.38 (m, 12H), 2.71 (m, 1H), 1.65 (m, 1H), 1.60 (s, 3H), 1.57 (m, 2H), 1.50 (s, 3H), 0.97 (s, 9H), 0.89 (d, J = 6.1 Hz, 3H), 0.88 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.2, 170.8, 170.3, 156.7, 136.7, 128.6, 128.2, 110.9, 74.7, 70.4, 70.3, 70.1, 69.7, 66.8, 60.9, 47.5, 41.0, 39.2, 36.9, 34.8, 26.9, 26.7, 25.9, 23.3, 22.0; HRMS (ESI) calcd. for C31H49N3O9Na+ (M + Na)+ 630.3367, found 630.3378.
:2 then 95:5) to afford methyl ester 8 (0.71 g, 83%) as a white foam. 1H NMR (300 MHz, CDCl3) δ 7.35–7.29 (m, 5H), 6.58 (br d, J = 6.7 Hz, 1H), 6.45 (m, 1H), 5.50 (m, 1H), 5.10 (s, 2H), 4.27–4.21 (m, 2H), 4.16 (br d, J = 6.8 Hz, 1H), 3.74 (s, 3H), 3.59–3.37 (m, 12H), 2.82 (m, 1H), 1.66–1.55 (m, 3H), 0.96 (s, 9H), 0.92 (d, J = 6.0 Hz, 3H), 0.91 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.7, 173.2, 170.1, 156.5, 136.4, 128.4, 128.0, 71.7, 70.1, 70.1, 69.9, 69.5, 66.5, 60.5, 52.3, 47.2, 40.7, 39.1, 38.4, 34.4, 26.5, 25.4, 22.4, 22.4; HRMS (ESI) calcd. for C29H47N3O9Na+ (M + Na)+ 604.3210, found 604.3212.
:4) to afford the title compound 10 (82 mg, 76% over 2 steps) as a white foam. 1H NMR (300 MHz, CDCl3) δ 7.21–7.17 (m, 8H), 7.09–7.05 (m, 12H), 6.94 (m, 1H), 6.80 (td, J = 8.8, 1.4 Hz, 1H), 6.56 (d, J = 9.3 Hz, 1H), 6.39 (m, 1H), 4.19–4.15 (m, 2H), 4.09 (d, J = 9.3 Hz, 1H), 3.73 (s, 3H), 3.71–3.33 (m, 12H), 2.77 (td, J = 7.7, 2.5 Hz, 1H), 1.66–1.51 (m, 3H), 0.93 (s, 9H), 0.92 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.7, 173.2, 170.0, 160.0, 141.8, 128.5, 127.3, 127.1, 96.9, 71.8, 70.3, 70.2, 69.6, 69.5, 60.6, 52.5, 47.3, 39.9, 39.1, 38.5, 34.5, 26.5, 25.6, 22.5, 22.4; 19F NMR (282 MHz, CDCl3) −16.5 (t, J = 11.3 Hz, 1F), −21.5 (s, 1F), −28.3 (dd, J = 16.9, 11.3 Hz, 1F); HRMS (ESI) calcd. for C54H61BN3O10F3Na+ (M + Na)+ 1002.4300, found 1002.4278.
:1, 0.8 mL) was added KCN (∼1 mg) followed by a solution of hydroxylamine in H2O (50 wt%, 100 μL, 1.5 mmol) and the reaction stirred at room temperature for 5.5 h. The mixture was then concentrated under reduced pressure and purified by preparative thin layer chromatography on silica gel (0.25 mm plate, CH2Cl2/MeOH, 93:7, twice) to afford the desired hydroxamic acid title compound 1 (9 mg, 30%) as a white oil. 1H NMR (300 MHz, CDCl3) δ 7.43 (m, 1H), 7.22–7.17 (m, 8H), 7.09–7.00 (m, 12H), 6.81 (br t, J = 10.1 Hz, 1H), 6.53 (m, 1H), 4.13–4.08 (m, 2H), 3.70–3.36 (m, 12H), 3.15–3.10 (m, 1H), 1.68–1.52 (m, 3H), 0.92 (s, 9H), 0.92–0.86 (m, 6H); 19F NMR (282 MHz, CDCl3) δ −16.2 (s, 1F), −21.4 (s, 1F), −27.8 (s, 1F); HRMS (ESI) calcd. for C53H60BN4O10F3Na+ (M + Na)+ 1003.4252, found 1003.4268.
For use in labeling, 1 as prepared above, was repurified by HPLC to remove residual silicates and other NMR-transparent contaminants: 2 mg of 1 in 100 μL of methanol and loaded onto an Agilent Eclipse XDB 5μ C18 300 Å 9.4mm × 250 mm semi-preparative column. Program: flow = 3 mL min−1, step gradient from A to B; (A = 0.1% TFA in water, B = 0.05% TFA in 100% MeCN) 0 to 2 min.: 10% B, linear gradient 2 to 5 min.: 10% B to 50%B, linear gradient 5 to 8 min: 50% B to 100% B, no gradient 8 to 11 min: 100% B, linear gradient 11 to 15 min: 100% B to 10% B, wash/re-equilibration: 15 min to 18 min: 10% B. Temp: 50 degrees, with desired product eluting 11.25–11.8 min. Average recovery is approximately ∼1mg (∼1 μmol), which is then aliquoted into PCR tubes in portions of 100 nmol, lyophilized, and stored at −20 degrees.
Acknowledgements
This research was supported by grants from the National Cancer Institute of Canada (with funds raised by the Canadian Cancer Association), a Special Program Grant from the Canadian Breast Cancer Research Alliance Metastasis, and a Centre Grant from the Michael Smith Research Foundation to the UBC Centre of Blood Research. Salary support for D.M.P. was provided by the Michael Smith Foundation for Health Research of British Columbia. C.M.O. is supported by a Canada Research Chair in Metalloproteinase Proteomics and Systems Biology. P.F.L. is supported through a Feodor Lynen Research Fellowship of the Alexander von Humboldt Foundation and the German Federal Ministry of Education and Research.Notes and references
- S. Lee, J. Xie and X. Y. Chen, Biochemistry, 2009, 49, 1364–1376 CrossRef.
- S. Liu, Bioconjugate Chem., 2009, 20, 2199–2213 CrossRef CAS.
- M. C. Lasne, C. Perrio, J. Rouden, L. Barre, D. Roeda, F. Dolle and C. Crouzel, Top. Curr. Chem., 2002, 222, 201–258 CrossRef CAS.
- S. A. Moerlein and J. S. Perlmutter, Eur. J. Pharmacol., 1992, 218, 109–115 CrossRef CAS.
- J. L. Lim, L. Zheng, M. S. Berridge and T. J. Tewson, Nucl. Med. Biol., 1996, 23, 911–915 CrossRef CAS.
- J. Becaud, L. J. Mu, M. Karramkam, P. A. Schubiger, S. M. Ametamey, K. Graham, T. Stellfeld, L. Lehmann, S. Borkowski, D. Berndorff, L. Dinkelborg, A. Srinivasan, R. Smits and B. Koksch, Bioconjugate Chem., 2009, 20, 2254–2261 CrossRef CAS.
- O. Jacobson, L. Zhu, Y. Ma, I. D. Weiss, X. Sun, G. Niu, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2011, 22, 422–428 CrossRef CAS.
- P. Laverman, W. J. McBride, R. M. Sharkey, A. Eek, L. Joosten, W. J. G. Oyen, D. M. Goldenberg and O. C. Boerman, J. Nucl. Med., 2010, 51, 454–461 CrossRef CAS.
- L. Iovkova, B. Wangler, E. Schirrmacher, R. Schirrmacher, G. Quandt, G. Boening, M. Schurmann and K. Jurkschat, Chem.–Eur. J., 2009, 15, 2140–2147 CrossRef CAS.
- R. Schirrmacher, G. Bradtmoller, E. Schirrmacher, O. Thews, J. Tillmanns, T. Siessmeier, H. G. Buchholz, P. Bartenstein, B. Waengler, C. M. Niemeyer and K. Jurkschat, Angew. Chem., Int. Ed., 2006, 45, 6047–6050 CrossRef CAS.
- J. Schulz, D. Vimont, T. Bordenave, D. James, J. M. Escudier, M. Allard, M. Szlosek-Pinaud and E. Fouquet, Chem.–Eur. J., 2011, 17, 3096–3100 CrossRef CAS.
- R. Ting, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2005, 127, 13094–13095 CrossRef CAS.
- J. M. Hooker, Curr. Opin. Chem. Biol., 2010, 14, 105–111 CrossRef CAS.
- S. Daniels, S. F. M. Tohid, W. Velanguparackel and A. D. Westwell, Expert Opinion on Drug Discovery, 5, pp. 291–304 Search PubMed.
- R. Ting, C. W. Harwig, U. auf dem Keller, S. McCormick, P. Austin, C. M. Overall, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2008, 130, 12045–12055 CrossRef CAS.
- P. Bohn, A. Deyine, R. Azzouz, L. Bailly, C. Fiol-Petit, L. Bischoff, C. Fruit, F. Marsais and P. Vera, Nucl. Med. Biol., 2009, 36, 895–905 CrossRef CAS.
- C. M. Overall and O. Kleifeld, Nat. Rev. Cancer, 2006, 6, 227–239 CrossRef CAS.
- C. M. Overall and O. Kleifeld, Br. J. Cancer, 2006, 94, 941–946 CrossRef CAS.
- C. M. Overall and C. López-Otín, Nat. Rev. Cancer, 2002, 2, 657–672 CrossRef CAS.
- U. A. D. Keller, C. L. Bellac, Y. Li, Y. M. Lou, P. F. Lange, R. Ting, C. Harwig, R. Kappelhoff, S. Dedhar, M. J. Adam, T. J. Ruth, F. Benard, D. M. Perrin and C. M. Overall, Cancer Res., 2010, 70, 7562–7569 CrossRef.
- S. Wagner, H. J. Breyholz, A. Faust, C. Holtke, B. Levkau, O. Schober, M. Schafers and K. Kopka, Curr. Med. Chem., 2006, 13, 2819–2838 CrossRef CAS.
- S. Wagner, H. J. Breyholz, M. P. Law, A. Faust, C. Holtke, S. Schroer, G. Haufe, B. Levkau, O. Schober, M. Schafers and K. Kopka, J. Med. Chem., 2007, 50, 5752–5764 CrossRef CAS.
- S. Wagner, H.-J. Breyholza, C. Höltkea, A. Fausta, O. Schobera, M. Schäfersa and K. Kopkaa, Appl. Radiat. Isot., 2009, 67, 606–610 CrossRef CAS.
- J. E. Sprague, W. P. Li, K. X. Liang, S. Achilefu and C. J. Anderson, Nucl. Med. Biol., 2006, 33, 227–237 CrossRef CAS.
- S. Furumoto, K. Takashima, K. Kubota, T. Ido, R. Iwata and H. Fukuda, Nucl. Med. Biol., 2003, 30, 119–125 CrossRef CAS.
- R. Ting, J. Lo, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Fluorine Chem., 2008, 129, 349–358 CrossRef CAS.
- B. Kuhnast, F. Hinnen, R. Hamzavi, R. Boisgard, B. Tavitian, P. E. Nielsen and F. Dollé, J. Labelled Compd. Radiopharm., 2005, 48, 51–61 CrossRef CAS.
- J. A. Inkster, M. J. Adam, T. Storr and T. J. Ruth, Nucleosides, Nucleotides Nucleic Acids, 2009, 28, 1131–1143 CAS.
- R. Ting, C. Harwig, J. Lo, Y. Li, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Org. Chem., 2008, 73, 4662–4670 CrossRef CAS.
- K. Jenssen, K. Sewald and N. Sewald, Bioconjugate Chem., 2004, 15, 594–600 CrossRef CAS.
- I. Stern and B. Shapiro, J. Clin. Pathol., 1953, 6, 158–160 CrossRef CAS.
- A. Antonis, J. Lipid Research-Notes on Methodology, 1960, 1, 485–486 CAS.
- R. E. Notari and J. W. Munson, J. Pharm. Sci., 1969, 58, 1060 CrossRef CAS.
- W. C. Eckelman, M. Bonardi and W. A. Volkert, Nucl. Med. Biol., 2008, 35, 523–527 CrossRef CAS.
- J. Bergman, O. Eskola, P. Lehikoinen and O. Solin, Appl. Radiat. Isot., 2001, 54, 927–933 CrossRef CAS.
- M. S. Berridge, S. M. Apana and J. M. Hersh, J. Labelled Compd. Radiopharm., 2009, 52, 543–548 CrossRef CAS.
- M. Glaser, H. Karlsen, M. Solbakken, J. Arukwe, F. Brady, S. K. Luthra and A. Cuthbertson, Bioconjugate Chem., 2004, 15, 1447–1453 CrossRef CAS.
- A. J. Beer, M. Niemeyer, J. Carlsen, M. Sarbia, J. Nahrig, P. Watzlowik, H.-J. Wester, N. Harbeck and M. Schwaiger, J. Nucl. Med., 2008, 49, 255–259 CrossRef.
- H. J. Wester, M. Schottelius, K. Scheidhauer, G. Meisetschlager, M. Herz, F. C. Rau, J. C. Reubi and M. Schwaiger, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 117–122 CrossRef CAS.
- C. J. Anderson, F. Dehdashti, P. D. Cutler, S. W. Schwarz, R. Laforest, L. A. Bass, J. S. Lewis and D. W. McCarthy, J. Nucl. Med., 2001, 42, 213–221 CAS.
- G. Ceccarini, R. R. Flavell, E. R. Butelman, M. Synan, T. E. Willnow, M. Bar-Dagan, S. J. Goldsmith, M. J. Kreek, P. Kothari, S. Vallabhajosula, T. W. Muir and J. M. Friedman, Cell Metab., 2009, 10, 148–159 CrossRef CAS.
- R. Haubner, H. J. Wester, W. A. Weber, C. Mang, S. I. Ziegler, S. L. Goodman, R. Senekowitsch-Schmidtke, H. Kessler and M. Schwaiger, Cancer Research, 2001, 61, 1781–1785 CAS.
- T. Ujula, M. Huttunen, P. Luoto, H. Perkyl, I. Simpura, I. Wilson, M. Bergman and A. Roivainen, Bioconjugate Chem., 2010, 21 Search PubMed.
- S. R. Banerjee, M. Pullambhatla, Y. Byun, S. Nimmagadda, G. Green, J. J. Fox, A. Horti, R. C. Mease and M. G. Pomper, J. Med. Chem., 2010, 53, 5333–5341 CrossRef CAS.
- Y. Chen, C. A. Foss, Y. Byun, S. Nimmagadda, M. Pullambahatla, J. J. Fox, M. Castanares, S. E. Lupold, J. W. Babich, R. C. Mease and M. G. Pomper, J. Med. Chem., 2008, 51, 7933–7943 CrossRef CAS.
- R. R. Flavell, P. Kothari, M. Bar-Dagan, M. Synan, S. Vallabhajosula, J. M. Friedman, T. W. Muir and G. Ceccarini, J. Am. Chem. Soc., 2008, 130, 9106–9112 CrossRef CAS.
- S. A. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS.
- M.-P. Kung and H. F. Kung, Nucl. Med. Biol., 2005, 32, 673–678 CrossRef CAS.
- K. J. Guenther, S. Yoganathan, R. Garofalo, T. Kawabata, T. Strack, R. Labiris, M. Dolovich, R. Chirakal and J. F. Valliant, J. Med. Chem., 2006, 49, 1466–1474 CrossRef CAS.
- W. A. P. Breeman, D. J. Kwekkeboom, P. P. M. Kooij, W. H. Bakker, L. J. Hofland, T. J. Visser, G. J. Ensing, S. W. J. Lamberts and E. P. Krenning, J. Nucl. Med., 1995, 36, 623–627 CAS.
- M. Namavari, O. P. De Jesus, Z. Cheng, A. De, E. Kovacs, J. Levi, R. Zhang, J. K. Hoerner, H. Grade, F. A. Syud and S. S. Gambhir, Mol. Imaging Biol., 2008, 10, 177–181 CrossRef.
- B. Kuhnast, F. Hinnen, R. Hamzavi, R. Boisgard, B. Tavitian, P. E. Nielsen and F. Dolle, J. Labelled Compd. Radiopharm., 2005, 48, 51–61 CrossRef CAS.
- A. M. Elizarov, R. M. van Dam, Y. S. Shin, H. C. Kolb, H. C. Padgett, D. Stout, J. Shu, J. Huang, A. Daridon and J. R. Heath, J. Nucl. Med., 2010, 51, 282–287 CrossRef CAS.
- C. C. Lee, G. D. Sui, A. Elizarov, C. Y. J. Shu, Y. S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake and H. R. Tseng, Science, 2005, 310, 1793–1796 CrossRef CAS.
- A. M. Elizarov, Lab Chip, 2009, 9, 1326–1333 RSC.
- C. R. Wade, H. Zhao and F. P. Gabbai, Chem. Commun., 2010, 46, 6380–6381 RSC.
- H. Ebersbach, E. Fiedler, T. Scheuermann, M. Fiedler, M. T. Stubbs, C. Reimann, G. Proetzel, R. Rudolph and U. Fiedler, J. Mol. Biol., 2007, 372, 172–185 CrossRef CAS.
- B. C. Roy and S. Mallik, J. Org. Chem., 1999, 64, 2969–2974 CrossRef CAS.
- R. J. Davenport and R. J. Watson, Tetrahedron Lett., 2000, 41, 7983–7986 CrossRef CAS.
- M. J. Fray and D. Ellis, Tetrahedron, 1998, 54, 13825–13832 CrossRef CAS.
- C. Y. Ho, E. Strobel, J. Ralbovsky and R. A. Galemmo, J. Org. Chem., 2005, 70, 4873–4875 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, HPLC traces and other important data. See DOI: 10.1039/c1md00117e |
| This journal is © The Royal Society of Chemistry 2011 |