DOI:
10.1039/C1MD00116G
(Concise Article)
Med. Chem. Commun., 2011,
2, 828-839
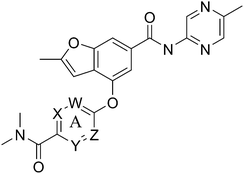
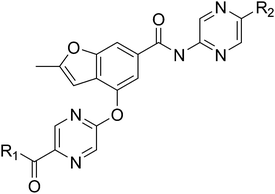
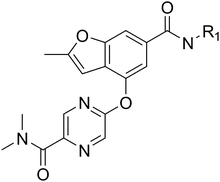
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus†
Received
4th May 2011
, Accepted 7th June 2011
First published on 5th July 2011
Abstract
Glucokinase is a key regulator of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
Introduction
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public health problem affecting over 220 million people worldwide.1 The disease is characterized by elevated fasting plasma COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose (FPG), insulin resistance, abnormally elevated hepatic glucose production and reduced glucose-stimulated insulin secretion (GSIS).2 While several classes of diabetic therapies are available for clinical use, there still remains a significant need for new therapies with improved efficacy and safety to help patients achieve their treatment goals.3
Glucokinase activators offer a promising opportunity for the treatment of T2DM patients.4Glucokinase (GK) is responsible for the conversion of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose to COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose-6-phosphate (G-6-P), and it functions as a key regulator of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose homeostasis.5 In the liver, GK regulates hepatic COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose utilization and output whereas in the pancreas it functions as a glucostat establishing the threshold for beta-cell glucose-stimulated insulin secretion. Glucokinase is also found in COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose sensing neurons of the ventromedial hypothalamus where it regulates the counter regulatory response (CRR) to hypoglycemia.6 Finally, glucokinase is reportedly expressed in the endocrine K and L cells where its function is less well characterized but may help regulate incretin release.7 Therapeutically, it is anticipated that activation of glucokinase in the liver and pancreas would be an effective strategy for lowering blood COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose by up regulating hepatic COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose utilization, down regulating hepatic COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose output and normalizing COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose stimulated insulin secretion.
Glucokinase is unique among the members of the hexokinase family given its low substrate affinity (Km ∼ 8 mM), positive substrate cooperativity and lack of product inhibition. As a monomeric enzyme, glucokinase achieves this cooperativity through equilibration between multiple protein conformations.8 In pioneering work, Grimsby and coworkers demonstrated that small molecule activators were capable of binding to glucokinase at an allosteric site 20 Å remote from the active site and influencing the enzyme's kinetic profile by modulating both Km for COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose (also known as S0.5) and Vmax.9 These efforts resulted in the identification of a phenylacetamide series of activators including clinical candidates 1 and 2 (piragliatin).10,11 Several structurally related phenylacetamide series, represented by 3 and 4, have also been reported.12,13 Moreover, a variety of other structurally diverse glucokinase activators have been identified, including aryl amides 514 and 615 as well as COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundbenzimidazole 7.16 These and other small molecule activators of glucokinase have been recently reviewed.17 Small molecule glucokinase activators have been shown to effectively lower blood COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose in a variety of diabetic animal models; moreover, several compounds including piragliatin (2) and MK-0941 (structure not disclosed) have advanced to clinical studies and were found to effectively lower fasting and post-prandial COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose in healthy subjects as well as T2DM patients.18,19 However, during both pre-clinical and clinical studies, hypoglycemia has been revealed as a key risk for this class.4,18,19 Managing this hypoglycemia risk may require dose titration and could limit therapeutic utility. Hence, herein we report on the structure–activity optimization of a series of substituted benzofurans designed as “partial activators” of glucokinase which, in preclinical studies, exhibit robust efficacy with reduced hypoglycemia risk.
In vitro screening strategy to identify activator profiles with reduced hypoglycemia risk
When initiating our program, we sought to develop a screening cascade that would enable correlation of an activator's intrinsic biochemical properties (i.e. changes in glucokinase Km and Vmax) against its in vivo efficacy and safety profile in order to identify activator profiles with optimized therapeutic indexes. As a first step, the potency and activation profile of individual activators were determined using recombinant human glucokinase enzyme in a biochemical assay monitoring the rate of glucose-6-phosphate formation using a G6PDH/NADP coupled assay.20 While many previously reported glucokinase assays measured overall activation relative to control at a single glucose concentration (typically reported as percentage maximum activation),21 we sought to isolate the effects of a given activator on glucokinase's Km for COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose and its Vmax to better understand how structural modifications of activators affected these individual properties. Similar to the method recently described by Bebernitz and coworkers,12 a matrix assay was utilized wherein dose response determinations for an activator (at 22 different activator concentrations from 0–100 μM) were conducted at 16 different glucose concentrations (ranging from 0 to 100 mM) as illustrated in Fig. 2a. Using a non-essential activator model (Fig. 2b), an activator's maximum effect on reducing glucokinase's Km for COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose was defined as α and it's maximum effect on the enzyme's Vmax was defined as β as illustrated in Fig. 2c and 2d, respectively.22 Values of α ranged from 0–1 and the lower the α value in this range the more substantially an activator reduced the enzyme's glucose Km. Values of β > 1 represented activator induced increases in the enzyme's Vmax whereas β < 1 represented the situation where an activator reduced the enzyme's Vmax. In cases where β = 1, an activator has no effect on the enzyme's Vmax. An activator's potency or EC50 was defined as the concentration affording a half maximal reduction in Km. Using this assay, the benchmark activators from Fig. 1 were profiled against human and rat glucokinase with the results summarized in Table 1. As shown, all activators demonstrated sub-micromolar potencies, and inter-species potency differences tended to be minimal (<4-fold). The activators had α values ranging from 0.04–0.10 suggesting a relatively consistent and substantial reduction in the enzyme's Km for COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose. Moreover, most compounds, with the exception of 4 and 7 in rat, had β > 1 suggesting that they also increased the enzyme's maximum velocity.
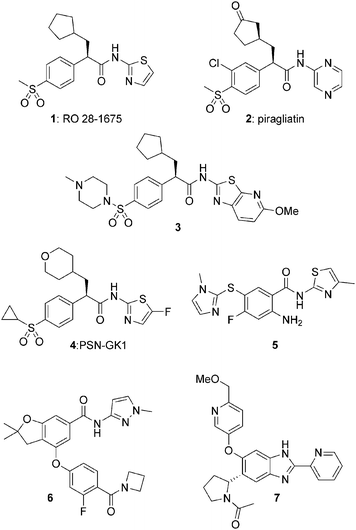 |
| | Fig. 1 Structures of representative glucokinase activators. | |
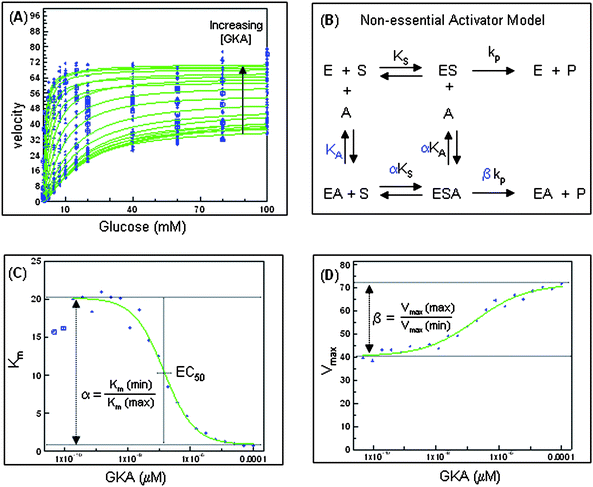 |
| | Fig. 2 Biochemical evaluation of prototypical glucokinase activator providing parameters EC50, α, and β. | |
Table 1 Biochemical activation profile of reference glucokinase activators 1–7a
| |
Human Glucokinaseb |
Rat Glucokinaseb |
| EC50 (nM) |
α |
β |
EC50 (nM) |
α |
β |
| See Fig. 1 for activator structures. Biochemical assay values reported as the mean of n ≥ 2 independent determinations unless otherwise noted. Data from n = 1 determination; NT = not tested. |
|
|
|
|
|
|
|
| 1 |
RO-28-1675 |
334 ± 194 |
0.10 ± 0.02 |
1.32 ± 0.11 |
235 ± 8 |
0.08 ± 0.02 |
1.16 ± 0.07 |
| 2 |
piragliatin |
364 ± 140 |
0.04 ± 0.01 |
1.73 ± 0.19 |
100 ± 13 |
0.04 ± 0.01 |
1.51 ± 0.09 |
| 3 |
|
122c |
0.04 |
1.40 |
390c |
0.05 |
1.50 |
| 4 |
PSN-GK1 |
37 ± 22 |
0.06 ± 0.01 |
1.22 ± 0.08 |
50c |
0.06 |
1.00 |
| 5 |
|
66 ± 32 |
0.04 ± 0.02 |
1.30 ± 0.06 |
NT |
NT |
NT |
| 6 |
|
27 ± 9 |
0.04 ± 0.01 |
1.29 ± 0.07 |
50 ± 5 |
0.05 ± 0.01 |
1.05 ± 0.09 |
| 7 |
|
88 ± 76 |
0.07 ± 0.01 |
1.08 ± 0.02 |
187 ± 73 |
0.09 ± 0.02 |
0.82 ± 0.02 |
Notably, despite the relative structural diversity of the activators described in Fig. 1, most offered similar activation profiles as shown in Table 1. However, a variety of alternative activation profiles could be postulated. For example, using the non-essential activator description, Fig. 3 illustrates simulations of various different hypothetical profiles (denoted as GKA-1 to GKA-4) on the glucose dependent velocity of glucokinase. These hypothetical profiles represent the effects of different α and β combinations assuming an activator concentration of 2-fold EC50. GKA-1 represents one extreme and is similar to activators such as 2 which induce a significant reduction in Km (indicated by low α) and a significant increase in Vmax (denoted by high β). The potential for COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose lowering efficacy of the GKA-1 profile in a hyperglycemic state (>7 mM COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose) is readily evident by the significant increase in enzyme velocity observed at high COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose relative to wild type enzyme; however, this same profile also results in significant increases in enzyme activity at low COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose potentially contributing to hypoglycemia risk. For perspective, the simulated enzyme velocity in the presence of GKA-1 (at 2-fold EC50) at 2.5 mM (45 mg/dL) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose approximately equals the velocity of wild type glucokinase at 15 mM (270 mg/dl) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose. It could be envisioned that activators with less dramatic reductions of Km (i.e. α values greater than GKA-1) combined with appropriate effects on Vmax might offer activation at high COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose (affording efficacy) but cause reduced activation at low COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose (avoiding hypoglycemia). Two potential examples of such “partial activators” are represented by GKA-2 and GKA-3. GKA-2 represents a modest reduction of Km (α = 0.5) coupled with an increase in Vmax (β = 1.5). Consistent with the suggestion of Bebernitz,12 such a profile would appear to be a favorable option for delivering efficacy in a hyperglycemic state while minimizing hypoglycemia risk. An additional option is represented by GKA-3 wherein the activator has an intermediate effect on Km (α = 0.1) and no effect on Vmax (β = 1.0). Relative to GKA-1, GKA-3 offers reduced activation at low COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose but still increases velocity at high COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose albeit not to the extent of GKA-1 or GKA-2. Hence, this profile may translate into lower hypoglycemic potential albeit with a risk of insufficient efficacy. A final hypothetical profile is represented by GKA-4 with an intermediate effect on Km (α = 0.1) and a suppression of Vmax (β = 0.5). As shown in Fig. 4, it would be anticipated that such a profile would be problematic as it would result in reduced enzyme velocity (relative to wild type) at high COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose and thus be non-efficacious, even if at low COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose it presents low hypoglycemia risk.
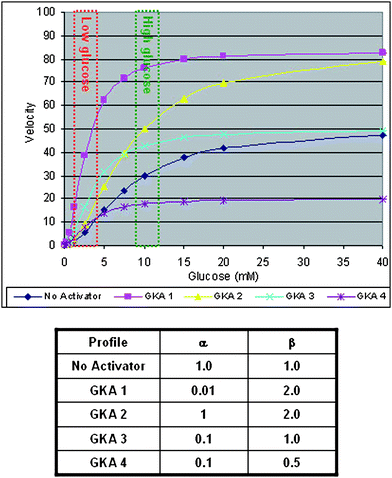 |
| | Fig. 3 Simulated affects of hypothetical activators (GKA-1, GKA-2, GKA-3 and GKA-4) on the glucose dependent velocity of glucokinase. | |
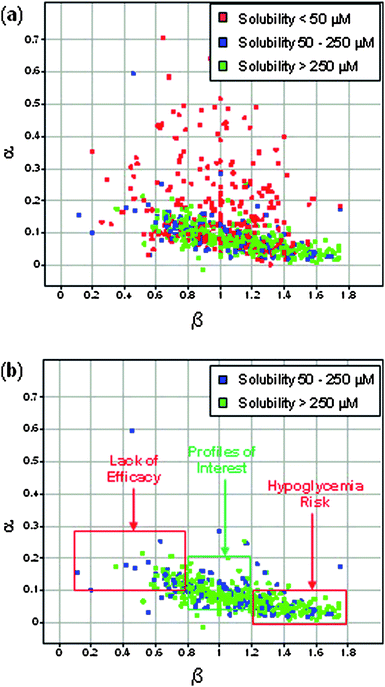 |
| | Fig. 4 (a) Plot of α vs. β for all compounds in screening set with EC50 < 10 μM; (b) Plot of α vs. β for all compounds in screening set with EC50 < 10 μM and kinetic solubility >50 μM. | |
To experimentally test whether alternative activator profiles such as GKA-2 and GKA-3 might offer an increased therapeutic index against hypoglycemia, we sought to experimentally identify activators representing these profiles and evaluate their in vivo efficacy and safety profiles relative to selected benchmarks from Table 1. A diverse screening set was constructed by combining internal HTS hits15 with structurally diverse glucokinase activators reported in the literature. This screening set was profiled in the matrix assay (Fig. 2) to determine α,β and EC50 values for individual compounds. Activators with EC50 < 10 uM were then plotted in Fig. 4 to examine relationships between α and β. The kinetic solubility of each activator (included in Fig. 4 as color code) was also determined as it was found to be an important factor for an accurate determination of the α and β values of a compound which required testing to up to 100 uM. Specifically, it was determined that for compounds with low solubility (<50 uM) an accurate determination of α was problematic so these compounds were removed from the analysis as shown in Fig. 4b thereby revealing a trend that higher α values tended to be associated with lower β values. Among the postulated hypothetical profiles described in Fig. 3, it became evident that the profile of hypothetical GKA-2 was not represented in this experimental data set. By contrast, the chemical space surrounding the profiles of GKA-1, GKA-3 and to a lesser extent GKA-4 were well populated. We sub-divided potential leads into three categories: (a) potential hypoglycemia risk for α < 0.1 and β > 1.2; (b) profile of interest for α = 0.05–0.2 and β = 0.8–1.2; and (c) lack of efficacy risk for α > 0.1 and β < 0.8. Among the compounds in the middle category, we selected the 2-methyl benzofuran template shown in Fig. 5 as the starting point for structure activity optimization with the goal of optimizing potency, activation profile and pharmacokinetic properties to enable an in vivo evaluation of this profile.23
Chemistry
The substituted 2-methyl benzofurans utilized in our structure activity studies were prepared as illustrated for the representative analogs shown in Schemes 1 and 2.
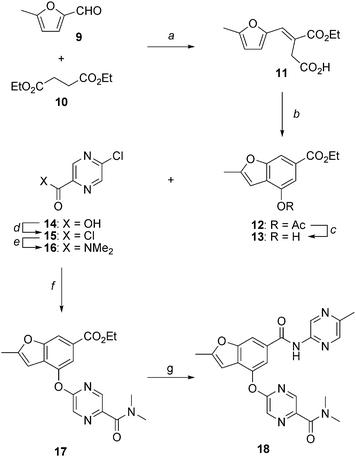 |
| | Scheme 1 Synthesis of 2-methylbenzofuran 18 as a glucokinase activator. Reagents and conditions: (a) NaOEt, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundEtOH, reflux, 13 h, 68%; (b) NaOAc, Ac2O, reflux, 2.5 h, 100%; (c) K2CO3, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundEtOH, 60 °C, 6 h, 90%; (d) (COCl)2, cat. DMF, CH2Cl2, 23 °C, 18 h, 100%; (e) Me2NH·HCl, Et3N, CH2Cl2, 23 °C, 4 h, 85%; (f) Cs2CO3, DMF, 90 °C, 3 h, 95%; (g) 2-amino-5-methylpyrazine, Me2AlCl, DME, reflux, 18 h, 72%. | |
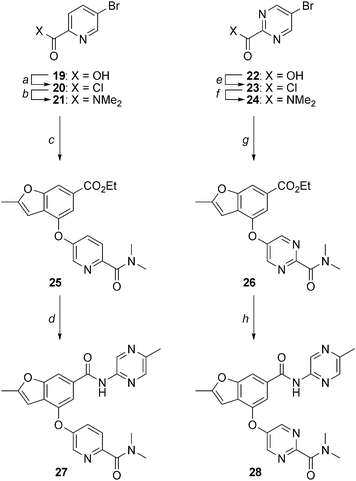 |
| | Scheme 2 Synthesis of 2-methylbenzofurans 27 and 28. Reagents and conditions: (a) (COCl)2, cat. DMF, CH2Cl2, 23 °C, 18 h; (b) Me2NH·HCl, Et3N, CH2Cl2, 23 °C, 4 h, 64% (two steps); (c) 13, Pd(OAc)2, tBuXPhos, K3PO4, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundtoluene, reflux, 24 h, 8%; (d) 2-amino-5-methylpyrazine, Me2AlCl, DME, reflux, 18 h, 23%; (e) (COCl)2, cat. DMF, DCM, 23 °C, 2 h, 100%; (f) 2M Me2NH in THF, THF, 23 °C, 16 h, 77%; (g) 13, CuI, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compound1,10-phenanthroline, Cs2CO3, DMF, 90 °C, 18 h, 54%; (h) 2-amino-5-methylpyrazine, Me2AlCl, DME, reflux, 3.5 h, 81%. | |
As shown in Scheme 1, base-mediated condensation of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compound5-methyl-2-furaldehyde (9) and COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddiethyl succinate (10) provided α,β-unsaturated intermediate 11 in 68% yield. Subsequent treatment of 11 with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundacetic anhydride and COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundsodium acetate at elevated temperature afforded cyclization to 2-methyl benzofuran 12 in quantitative yield. The acetate of 12 was removed by treatment with K2CO3 in the presence of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundEtOH to provide COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundphenol 13.24 An alternative process scale synthesis of 13 has also been reported.25 In preparation for coupling of the lower heteroaryl ring, 5-chloropyrazine-2-carboxylic acid (14) was converted to the corresponding acid chloride 15 and then reacted with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddimethylamine to generate amide 16 in 85% yield. An aromatic nucleophilic substitution reaction between 13 and 16 in the presence of cesium carbonate at elevated temperature afforded heteroaryl ether 17 in 95% yield. Finally, transamidation of ester 17 with 5-methyl-2-aminopyrazine in the presence of dimethylaluminum chloride afforded amide 18 in 72% yield. Utilization of alternative coupling partners in the nucleophilic aromatic substitution and transamidation reactions enabled preparation of a variety of structurally diverse analogs as described in the experimental section.
For analogs wherein the lower aryl/heteroaryl ring was not amenable to installation via an aromatic nucleophilic substitution reaction, palladium- or copper-mediated coupling methods were employed as highlighted in Scheme 2. For example, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundpyridine analog 27 was made from 5-bromopyridine-2-carboxylic acid (19) which was converted to the corresponding acid chloride 20 and reacted with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddimethylamine to generate amide 21. Treatment of 21 with phenol 13 in the presence of catalytic Pd(OAc)2, tBuXPhos and K3PO4 in COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundtoluene at reflux resulted in conversion to aryl ether 25 in 8% yield.26 Transamidation of the ester of 25 with 5-methyl-2-aminopyrazine in the presence of dimethylaluminum chloride afforded amide 27. Representative pyrimidine analog 28 was prepared from 5-bromopyrimidine-2-carboxylic acid (22) which was converted to the corresponding acid chloride 23 and reacted with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddimethylamine to generate amide 24. Treatment of 24 with phenol 13 in the presence of catalytic COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compound1,10-phenanthroline and COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundcopper iodide in COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddimethylformamide at 90 °C resulted in conversion to aryl ether 26 in 54% yield.27 Finally, transamidation of the ester of 26 with 5-methyl-2-aminopyrazine in the presence of dimethylaluminum chloride afforded amide 28.
Structure activity relationships
All analogs were initially evaluated in the biochemical activation assay (Fig. 2) measuring potency (EC50) as well as effects on Km (α) and Vmax (β). Additionally, human liver microsome (HLM) stability, passive permeability, kinetic solubility and COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddofetilide binding, as a surrogate for hERG inhibitory activity, were evaluated for all analogs. The structure activity trends are summarized in Tables 2–5.
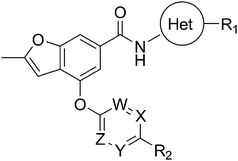
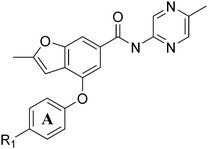
Table 2 Biochemical potency, activation profile and ADME properties of 29–32
| 
|
| |
R 1 |
Biochemical Activationa |
HLM CIint (ml min−1 kg−1) |
Permeabilityb (10−6 cm s−1) |
Kinetic Solubility (μM) |
COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDofetilide Binding IC50 (μM) |
| EC50 (nM) |
α |
β |
| Biochemical assay values reported as the mean ± SD of n ≥ 2 independent determinations unless otherwise noted. Passive permeability assessed in RRCK cell line. Data from n = 1 determination; NT = not tested. |
|
|
|
|
|
|
|
|
| 29 |
Me |
6610 ± 4156 |
0.23 ± 0.06 |
1.04 ± 0.09 |
47 |
0.3 (L) |
18 |
NT |
| 30 |
SO2Me |
870 ± 450 |
0.07 ± 0.01 |
1.27 ± 0.05 |
13 |
7.6 (M) |
3 |
22 |
| 31 |
CN |
3061 ± 974 |
0.22 ± 0.04 |
1.25 ± 0.03 |
29 |
1.5 (L) |
2 |
4 |
| 32 |
C(O)NMe2 |
92c |
0.10 |
0.69 |
17 |
5.9 (M) |
NT |
59 |
Table 3 Biochemical potency, activation profile and ADME properties of 18, 27–28 and 32–33
Table 4 Biochemical potency, activation profile and ADME properties of 18 and 34–43
Table 5 Biochemical potency, activation profile and ADME properties of 18 and 44–50
As shown in Table 2, initial structure–activity studies focused on an evaluation of the A-ring substitution (i.e. R1). Preliminary data (not shown) revealed that substitution in the para position of the A-ring provided optimal activity. Subsequent evaluation of a representative set of R1 substituents revealed that polar groups including R1 = SO2Me (30), R1 = CN (31) and R1 = C(O)NMe2 (32) offered improved potency and metabolic stability relative to more lipophilic substituents such as R1 = Me (29). In particular, the tertiary amide 32 offered optimal potency along with a maximum Kmreduction (α = 0.10) in the desired range; however, 32 also caused an undesirable suppression of Vmax (β = 0.69) and exhibited moderate COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddofetilide binding which needed to be resolved through subsequent optimization.
Modification of the A-ring of 32 explored both heterocyclic replacements (Table 3) and alternative amide substituents (Table 4). As shown in Table 3, replacement of the phenyl A-ring of 32 with various 6-membered heterocycles reduced lipophilicity affording improved metabolic stability, favorable solubility and reduced COMPOUND LINKS
Read more about this on ChemSpider
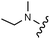
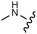
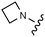
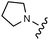
Download mol file of compounddofetilide binding. Somewhat unexpectedly these more polar analogs also had improved passive permeability relative to 32. This increased permeability measurement may perhaps be an indirect result of the improved solubility of these heterocycles in the RRCK assay system. Whereas the pyridine 27 resulted in a 5-fold loss of potency relative to 32, pyrazine 18 and pyrimidine 28 were only two-fold less potent and had activation profiles of interest: 18 (EC50 = 212 nM, α = 0.09, β = 0.98) and pyrimidine 27 (EC50 = 188 nM, α = 0.10, β = 0.87) offered the most favorable combinations of potency and activation profile. The regioisomeric pyrimidine 33 was inactive. In subsequent studies, the A-ring pyrazine of 18 was held constant, and a variety of secondary and tertiary amides were evaluated as shown in Table 4. While secondary amides (e.g.35) were significantly less active than the dimethyl tertiary amide of 18, cyclic tertiary amides, such as 36 (EC50 = 85 nM) and 37 (EC50 = 64 nM) offered improved potency relative to the acyclic amides. A 4–5 membered ring appears to be ideal since a larger ring size as in 38 resulted in a slight loss in potency and an increase in metabolic turnover. Unfortunately, despite their promising potency, these cyclic amides had poor aqueous solubilities and tended to have an inhibitory effect on Vmax (37: β = 0.67) particularly with ring sizes ≥5. In an effort to capitalize on the potency of these cyclic amides various substituents were explored (39–43) with the goal of increasing solubility and β.28 While these substituents generally afforded improved solubility and in some cases also increased β (e.g.43), these improvements came with significant losses in potency relative to the parent compound (i.e.36). Based on these observations, the original dimethyl amide of 18 was maintained in subsequent structure–activity studies given its favorable balance of potency, activation profile and physical properties.
A final region of structure–activity studies was the heterocyclic amide (R1) as shown in Table 5 evaluating variously substituted pyridines, pyrazines, pyrazoles and pyrimidines. As illustrated, the 5-methyl pyrazine (18) was found to be slightly more potent than the corresponding pyridine (44) or the related N-methyl pyrazole (49). The pyrimidine 50 was inactive. Within the pyrazine series, substitution at the 5-position tended to offer increased potency relative to the unsubstituted heterocycle (i.e.18, 46, 47, 48 all > 45), and among the analogs in Table 5, 5-methyl pyrazine 18 tended to offer the most optimal balance of potency, profile, metabolic stability and solubility. Based on these structure activity studies, compounds 18 and 28 were identified as offering optimal combinations of potency, activation profile, metabolic stability and solubility. Activator 28 was ultimately selected for additional characterization (vide infra) based on favorable biopharmaceutical properties relative to 18.
Structural biology studies
To understand the molecular interaction of 28 with the allosteric site of glucokinase, structural biology studies were conducted using glucokinase protein expressed in E.coli and purified as previously reported.9 Experimentally, glucokinase protein in buffer containing 25 mM HEPES pH 7.0, 0.5 mM TCEP, 0.05 M NaCl, and 40 mM COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose and 1 mM 28 at 20 mg ml−1 was used in hanging drop vapor diffusion crystallization over wells containing 0.1 M TrisHCl pH 7.0, 80–200 mM COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose, and 19–26% PEG-4000. X-ray crystallography data was collected at 17ID IMCA beamline at APS at 100 K, and the structure was determined using molecular replacement method using existing models. Coordinates for this structure have been deposited into the PDB and are available referencing 3S41. Examining the co-crystal structure in Fig. 6, the benzene of the benzofuran of 28 positions at the center of the same allosteric site reported before. The COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compound5-methylpyrazine, the dimethyl pyrimidine carboxamide and the methyl furan groups extend from this central benzene to three sub binding sites. The COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundmethylpyrazine is located in a tunnel formed by helix-C (residues 442-C-terminal) and loop 64–72, forming two conserved hydrogen bonds with main chain atoms of R63 on the loop. The COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundmethylfuran occupies a hydrophobic pocket formed by the side chain of M235, loop 64–71, Y214 and I211. The COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddimethyl pyrimidine carboxamide interacts with the side chain of Y215, helix-C as well as V91 and V101 from the mobile loop 91–101. Studies using solution-based methods further define the impact of 28 on the equilibrium between various GK conformations will be reported separately.
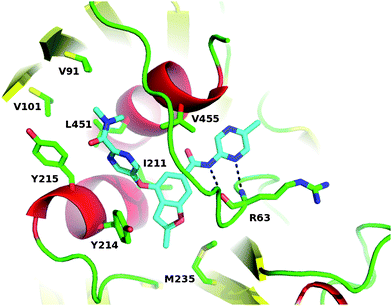 |
| | Fig. 6 Co-crystal structure of 28 bound to the allosteric site of human glucokinase. | |
Preclinical pharmacokinetics
The pharmacokinetic properties of 28 were characterized in rat, dog and monkey as summarized in Table 6. Compound 28 was found to have low clearance in dog and moderate clearance in rat and monkey. The compound had moderate oral bioavailability in rat and monkey and low bioavailability in dog. A slight gender difference was observed in rats. After IV dose of 28 in rats at 1 mg kg−1, 0.26% of the dose was excreted in urine as parent suggesting negligible renal clearance. After IV dose of 28 in bile duct-cannulated rats at 5 mg kg−1, 2.3% of the dose was excreted in bile as parent indicating that biliary excretion is not a major route of excretion in rat. Rat and dog liver microsomes were found to predict the rat and dog in vivo clearance, respectively, suggesting that the main route of clearance is cytochrome P450 mediated.
Table 6 Preclinical Pharmacokinetics of 28a
| Species |
Gender |
Cl (mL min−1 kg−1) |
T1/2 (hr) |
Vdss (L kg−1) |
%F |
| Pharmacokinetic parameters expressed as geometric mean of n = 2 animals per group. Spague-Dawley rats dosed 1.0 mg kg−1i.v. and mg kg−1p.o. Beagle dogs dosed at 0.5 mg kg−1i.v. and 5 mg kg−1p.o. Cynomolgus monkeys dosed at 0.5 mg kg−1i.v. and 3 mg kg−1p.o. |
|
|
|
|
|
| Rat |
M |
24.8 |
4.3 |
2.1 |
23% |
| Rat |
F |
13.0 |
1.2 |
1.1 |
77% |
| Dog |
M |
2.6 |
3.3 |
0.65 |
12% |
| Monkey |
M |
9.4 |
1.8 |
0.83 |
78% |
Activity in dispersed rat islets and hepatocytes
In the pancreatic β-cell, glucokinase serves as a glucostat establishing the threshold for glucose-stimulated insulin secretion.5 The effect of 28 on glucose-stimulated insulin secretion was evaluated in dispersed rat islets at concentrations of 0.4, 1.2, 4.0 and 12 μM which corresponded to a 1-, 3-, 10- and 30-fold coverage, respectively, of its EC50 against rat glucokinase (407 nM), as shown in Fig. 7. Compound 28 was tested for its ability to shift the threshold for glucose-stimulated insulin secretion in rat dispersed islet static culture and was found to increase insulin secretion at glucose concentrations of 9, 11, and 13 mM COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose with no effect at concentrations above 22 mM. When tested in a range 1 or 3 times the biochemical EC50, there was no effect on insulin secretion at low glucose concentrations of 3 and 5 mM, with effects only seen at compound concentrations of 10–30 times the EC50 (Fig. 7).
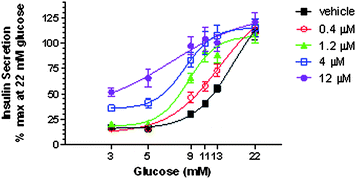 |
| | Fig. 7 Threshold shift of glucose-stimulated insulin secretion in rat dispersed islet static culture following administration of 28 at 0.4, 1.2, 4.0 and 12 μM. Data are expressed as Means ± Standard Error. | |
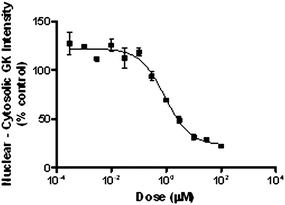
In contrast to pancreatic β-cells, glucokinase activity in hepatocytes is regulated though an interaction with glucokinase regulatory protein (GKRP).29 Physiologically, in a low glucose state, GKRP binds the inactive conformation of glucokinase and sequesters the enzyme to the nucleus. As intracellular glucose concentrations increase, glucokinase is released from GKRP and diffuses into the cytoplasm. Since small molecule GK activators exert their effects by altering the conformation of GK, these activators have been shown to disrupt the GK-GKRP interaction. To characterize the effects of 28 on the sub-cellular localization of GK in hepatocytes, a dose response evaluation was conducted in cryopreserved hepatocytes at 8.9 mM COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose and glucokinase translocation from the nucleus to the cytoplasm was monitored via fluorescence based cellomics analysis. Fig. 8 illustrates the dose dependent effect of 28 on translocation of GK from the nucleus to the cytoplasm with EC50 = 0.9 μM. Similar experiments were repeated at 2.5, 5.5 and 15 mM COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose affording EC50 values of 3.8, 2.3 and 0.6 μM, respectively.
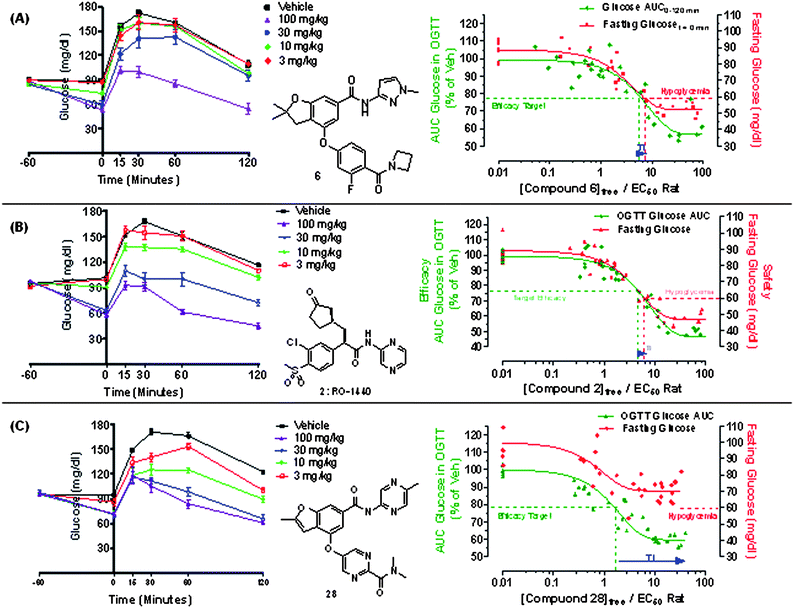
In vivo efficacy and hypoglycemia safety
To test the hypothesis that a glucokinase activator with the profile of 28 might offer a favorable therapeutic index relative to previous benchmark activators, we utilized an acute in vivo model to conduct a pharmacokinetic/pharmacodynamic (PK/PD) assessment of post-prandial efficacy and hypoglycemia risk. Specifically, as shown in Fig. 9, an oral glucose tolerance test (OGTT) was conducted in Sprague-Dawley rats wherein animals were fasted overnight and then orally dosed with the glucokinase activator at t = 0 min. At t = 60 min post dose, the effect of the activator on reducing fasting plasma COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose was determined as a preliminary measure of hypoglycemia safety (i.e. the propensity of an activator to lower COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose from a euglycemic to a hypoglycemic level). In these studies, hypoglycemia was defined as a fasting plasma COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose level of <60 mg/dl. To evaluate efficacy, an oral glucose challenge (2 g kg−1) was then administered 60 min post dose and the effect of the activator at reducing the glucose AUC excursion during a hyperglycemic state was determined. The predicted Ceff was defined as the free concentration of the GKA that reduced the excursion AUC by 20–25% (indicated at 23% on graph) during the OGTT excursion. An illustration of this experiment is shown for benchmark compound 6 as shown in Fig. 9A where p.o. dosing (3–100 mg kg−1) resulted in a dose-dependent reduction in fasting COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose over the first hour (t = −60 to 0 min) with higher doses reaching hypoglycemic levels (i.e. <60 mg/dL). During the subsequent OGTT (t = 0 to 120 min), compound 6 was efficacious and dose dependently reduced the post-prandial excursion. Notably, compound 6 starts displaying robust efficacy at a dose of 30 mg kg−1, at which hypoglycemia is also evident at the 60 min time point. The window between efficacy and hypoglycemia for compound 6 is further illustrated by plotting the PK/PD exposure effect relationship for the activator's effect both on fasting and post-prandial COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose. In this graph, the relationship of free drug concentration (normalized by rat EC50) versus fasting COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose (red line) and post-prandial COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose (green line) is indicated. As shown, the concentration of 6 that affords an efficacious reduction is very near the concentration resulting in lowering of fasting COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose to a hypoglycemic level. This observation of narrow pre-clinical therapeutic index in this model was not limited to 6 as evaluation of structurally distinct 2 (Fig. 9B) afforded a similarly narrow therapeutic index. This challenge model was subsequently utilized to evaluate compound 28 as shown in Fig. 9C. Oral dosing (3–100 mg kg−1) resulted in a dose-dependent reduction in post-prandial COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose during the OGTT. Although the maximum reduction of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose AUC with compound 28 seems to be slightly less than that with compounds 2 and 6, it appears to be sufficiently robust in this model. Importantly, while 28 also caused a reduction in fasting plasma COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglucose, it was not as substantial as that observed previously with activators 2 or 6 (Fig. 9A–9B) offering an improved preclinical therapeutic index as illustrated on the corresponding PK/PD plot in Fig. 9C.
The behavior of compound 28 in this model is consistent with the initial hypothesis that a glucokinase “partial activator” with a reduced effect on the enzyme's Km and little effect on the Vmax (GKA-3 profile in Fig. 3) could afford a lower risk of hypoglycemia than compounds such as 2 and 6 which have strong effects on both Km and Vmax (GKA-1 profile in Fig. 3). The efficacy of 28 has been further characterized in additional preclinical diabetic disease models, the results of which will be reported separately. While the efficacy and hypoglycemia data from in vivo preclinical models may not directly translate to diabetic patients due to underlying differences in physiology and pathophysiology, we postulate that activators such as 28 that are more efficacious in a hyperglycemic state and less effective in a euglycemic state are likely to afford improvements in the hypoglycemia safety of glucokinase activators in the clinic. Based on the promising efficacy and preclinical safety data, 28 was selected as an early development candidate and advanced to Phase 1 ascending single dose studies in Type 2 diabetic patients on stable COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundmetformin background therapy.
Notes and references
- World Health Organization Diabetes statistics. http://www.who.int/mediacentre/factsheets/fs312/en/ Accessed February 17, 2011.
- P. H. Bennett, W. C. Knowler, Definitions, Diagnosis and Classifications of Diabetes Mellitus and Glucose Homeostasis. In Joslin's Diabetes Mellitus, 14th ed. 331–339 Search PubMed.
- For a review, see: J. Xu, IDrugs, 2004, 7, 249 CAS.
- For reviews, see:
(a) M. C. T. Fyfe and M. J. Procter, Drugs Future, 2009, 34, 641 CAS;
(b) F. Matschinsky, Nat. Rev. Drug Discovery, 2009, 8, 399 CrossRef CAS.
- F. Matschinsky, M. A. Magnuson, ed. Glucokinase and Glycemic Disease: From Basics to Novel Therapeutics. Basel, 2004 Search PubMed.
- A. A. Dunn-Meynell, V. H. Routh, L. Kang, L. Gaspers and B. E. Levin, Diabetes, 2002, 51, 2056 CrossRef CAS.
- F. Reimann and F. M. Gribble, Diabetes, 2002, 51, 2757 CrossRef CAS.
- For a review, see: P. B. Iynedjian, Cell. Mol. Life Sci., 2008, 66, 27 CrossRef.
- J. Grimsby, R. Sarabu, W. L. Corbett, N. E. Haynes, F. T. Bizzarro, J. W. Coffey, K. R. Guertin, D. W. Hilliard, R. F. Kester, P. E. Mahaney, L. Marcus, L. Qi, C. L. Spence, J. Tengi, M. A. Magnuson, C. A. Chu, M. T. Dvorozniak, F. M. Matschinsky and J. F. Grippo, Science, 2003, 301, 370 CrossRef CAS.
- N. E. Haynes, W. L. Corbett, F. T. Bizzarro, K. R. Guertin, D. W. Hilliard, G. W. Holland, R. F. Kester, P. E. Mahaney, L. Qi, C. L. Spence, J. Tong, M. T. Dvorozniak, A. Railkar, F. M. Matschinsky, J. F. Grippo, J. Grimsby and R. Sarabu, J. Med. Chem., 2010, 53, 3618 CrossRef CAS.
- R. F. Kester, W. L. Corbett, R. Sarabu, P. E. Mahney, N.-E. Haynes, K. R. Guertin, F. T. Bizzarro, D. W. Hilliard, L. Qi, J. Tengi, J. F. Grippo, J. Grimsby, L. Marcus, C. Spence, M. Dvorozniak, J. Racha, K. Wang, 238th ACS national Meeting, August 16–20, 2009, Washington, DC.
- G. R. Bebernitz, V. Beaulieu, B. A. Dale, R. Deacon, A. Duttaroy, J. Gao, M. S. Grondine, R. C. Gupta, M. Kakmak, M. Kavana, L. C. Kirman, J. Liang, W. M. Maniara, S. Munshi, S. S. Nadkarni, H. F. Schuster, T. Stams, I. St. Denny, P. M. Taslimi, B. Vash and S. L. Caplan, J. Med. Chem., 2009, 52, 6142 CrossRef CAS.
- M. C. Fyfe, J. R. White, A. Taylor, R. Chatfield, E. Wargent, R. L. Printz, T. Sulpice, J. G. McCormack, M. J. Procter, C. Reynet, P. S. Widdowson and P. Wong-Kai-In, Diabetologia, 2007, 50, 1277 CrossRef CAS.
-
(a) T. T. Nishimura, T. Iino, Y. Nagata, J. Eiki, WO 2003080585 A1;
(b) K. Kamata, M. Mitsuya, T. Nishimura, J. Eiki and Y. Nagata, Structure, 2004, 12, 429 CrossRef CAS.
- S. Bailey, 12th Annual San Diego Med Chem Symposium, July 17, 2009; San Diego, CA.
- A. Klapars, K. R. Campos, J. H. Waldman, D. Zewge, P. G. Dormer and C. Chen, J. Org. Chem., 2008, 73, 4986 CrossRef CAS.
- For a reviews of reported glucokinase activators, see:
(a) M. Coghlan and B. Leighton, Expert Opin. Invest. Drugs, 2008, 17, 145 CrossRef CAS;
(b) R. Sarabu, S. J. Berthel, R. F. Kester and J. W. Tilley, Expert Opin. Ther. Pat., 2008, 18, 759 CrossRef CAS;
(c) R. Sarabu, S. J. Berthel, R. F. Kester and J. W. Tilley, Expert Opin. Ther. Pat., 2011, 21, 13 CrossRef CAS.
-
(a) J. Zhi, S. Zhai, M.-E. Mulligan, J. Grimsby, C. Arbet-Engels, M. Boldrin, R. Balena, Presentation 42, 2008 EASD 44th Annual Meeting, Rome, Italy;
(b) R. C. Bonadonna, T. Heise, C. Arbet-Engels, C. Kapitza, A. Avogaro, J. Grimsby, J. Zhi, J. F. Grippo and R. Balena, J. Clin. Endocrinol. Metab., 2010, 95, 5028 CrossRef CAS.
- Y. Shentu, Y. Chen, Q. Yu, D. Sheng, B. J. Musser, M. Alba, B. B. Zhang, E. M. Migoya, J. Ehrhart, K. D. Kaufman, B. J. Goldstein, EASD 46th Annual Meeting, September 20–24, 2010, Stockholm, Sweden.
- K. J. Brocklehurst, V. A. Payne, R. A. Davies, D. Carroll, H. L. Vertigan, H. J. Wightman, S. Aiston, I. D. Waddell, B. Leighton, M. P. Coghlan and L. Agius, Diabetes, 2004, 53, 535 CrossRef CAS.
- For a representative example of a glucokinase percent maximum activation assay, see: J. A. Pfefferkorn, J. Lou, M. L. Minich, K. J. Filipski, M. He, R. Zhou, S. Ahmed, J. Benbow, A. Guzman-Perez, M. Tu, J. Litchfield, R. Sharma, K. Metzler, F. Bourbonais, C. Huang, D. A. Beebe and P. J. Oates, Bioorg. Med. Chem. Lett., 2009, 19, 3247 CrossRef CAS.
- I. H. Segel. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems, Wiley, New York 1993 Search PubMed.
- For structurally related glucokinase activators based on aryl or heteraryl templates, see examples in ref. 17b and 17c.
-
(a) D. Simoni, R. Romagnoli, R. Baruchello, R. Rondanin, M. Rizzi, M. G. Pavani, D. Alloatti, G. Giannini, M. Marcellini, T. Riccioni, M. Castorina, M. B. Guglielmi, F. Bucci, P. Carminati and C. Pisano, J. Med. Chem., 2006, 49, 3143 CrossRef CAS;
(b) T. T. Howard, B. M. Lingerfelt, B. L. Purnell, A. E. Scott, C. A. Price, H. M. Townes, L. McNulty, H. L. Handl, K. Summerville, S. J. Hudson, J. P. Bowen, K. Kiakos, J. A. Hartley and M. Lee, Bioorg. Med. Chem., 2002, 10, 2941 CrossRef CAS.
- M. A. Berliner, E. M. Cordi, J. R. Dunetz and K. E. Price, Org. Process Res. Dev., 2010, 14, 180 CrossRef CAS.
- C. H. Burgos, T. E. Barder, X. Huang and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 4321 CrossRef CAS.
- P. J. Fagen, E. Hauptman, R. Shapiro and A. Casalnuovo, J. Am. Chem. Soc., 2000, 122, 5043 CrossRef.
- For synthesis of spirocyclic COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundoxetane of 43, see: G. Wuitshik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Marki, T. Godel, H. Fischer, B. Wagner, I. Parrillia, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Muller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512 CrossRef. - E. Van Schaftingen, A. Vandercammen, M. Detheux and D. R. Davies, Adv. Enzyme Regul., 1992, 32, 133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00116g |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.