Overcoming retinoic acid receptor-α based testicular toxicity in the optimisation of glucokinase activators†
Michael J.
Waring
*a,
Iain J.
Brogan‡
b,
Matthew
Coghlan
a,
Craig
Johnstone‡
a,
Huw B.
Jones
b,
Brendan
Leighton
a,
Darren
McKerrecher
a,
Kurt G.
Pike
a and
Graeme R.
Robb
a
aCardiovascular and Gastrointestinal Innovative Medicines Unit, AstraZeneca R&D, Mereside, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK. E-mail: mike.j.waring@astrazeneca.com; Tel: +44 (0) 1625 230942
bGlobal Safety Assessment, AstraZeneca R&D, Mereside, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK
First published on 10th June 2011
Abstract
Small molecule activators of the glucokinase enzyme have the potential to deliver a level of glycaemic control that is superior to current oral agents and hence have great promise as new therapies for Type 2 Diabetes. As such, attempts to discover glucokinase activators suitable for clinical development have been the focus of many major pharmaceutical research programmes. Here we describe how we have overcome a testicular toxicological liability in our pyridine acid lead series, which we show can be ascribed to antagonism of the retinoic acid receptor-α.
Introduction
Type 2 Diabetes (T2D) is a complex disease which affects 150 million people worldwide, with its prevalence expected to double by the year 2025.1 Current therapies do not achieve adequate glycaemic control2 and consequently there is an unmet need for therapeutic agents which deliver superior efficacy.Glucokinase (GK, also known as hexokinase IV or D) is expressed predominantly in the liver, pancreas, brain and gut where its function is to catalyse the conversion of glucose to glucose-6-phosphate. In both the liver, where the action of GK promotes glycogen synthesis, and the pancreas, where the action of GK results in glucose-sensitive insulin release,3GK is the rate limiting enzyme in glucose utilisation.4Activation of GK is therefore expected to improve glycaemic control by modulating hepatic glucose balance and decreasing the threshold for insulin secretion.5–7
Several groups have described small molecule glucokinase activators (GKAs) which act by binding to an allosteric site.8 We have described previously the identification and subsequent optimization of our own series of pyridine-5-carboxylic acid containing GKAs.9–11 This work resulted in the identification of GKA50 and GKA60, which showed excellent in vitro profiles, pharmacokinetic parameters and in vivo efficacy (Fig. 1). In addition, we have previously described how knowledge of the plasma compound levels following an oral dose, together with knowledge of enzyme potency and plasma protein binding, can be used to predict in vivo efficacy in an acute animal model.12
 | ||
| Fig. 1 The structures of pyridine-5-carboxylic acid containing GKAs. | ||
Results and discussion
Our intention was to progress GKA50 and GKA60 as clinical candidates but their progress was halted when it was revealed that both compounds cause epididymal and testicular toxicity in both rat and dog toxicology studies; namely exfoliated seminiferous epithelial cells and reduced spermatozoa (epididymes) and Sertoli cell vacuolation, spermatid retention and mild multifocal epithelial cell degeneration (testes) (Fig. 2), thus preventing progression of the compounds. It was necessary to explore whether these findings were linked inherently to activation of glucokinaseper se or inexorably to the chemical series. In order to address this, the toxicology profiles of two related compounds 1 and 2 were examined. These compounds were found to cause similar pathology in the testes at doses below those required for glucose lowering efficacy, suggesting that activation of glucokinase was not the cause of the problem. | ||
| Fig. 2 Photomicrographs of testis from control (A. ×20 and B. ×40 objective lens magnifications) and GKA50 treated animals (C. ×20 and D. ×40 objective lens magnifications) stained with haematoxylin and eosin. Normal seminiferous tubular morphology is present in controls and compares with substantial histopathological changes in compound 1 treated testes which show seminiferous tubular vacuolation (of Sertoli cells) leading to exfoliation of germ cells from the spermatogenic epithelium and substantial spermatid retention (indicated by the arrows) close to the Sertoli cell nuclear layer close to the tubular basal lamina. | ||
This information led us to hypothesise that the pathology was a consequence of alternative pharmacology present in these compounds. Hence, we decided to investigate whether structural variations would identify divergent structure–activity relationships between GK activation and the unwanted pathology. Examination of the substructures suggested that the pyridine acid motif was the most likely culprit since this is the only substructure common to all four compounds. Hence, we postulated that compounds without the carboxylic acid functionality might be free of the toxicity.
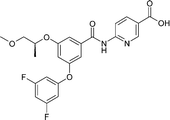
Replacement of the pyridine acid with certain neutral heterocycles was found to be tolerated in terms of GK potency. For instance, the thiazole derivative 4 was found to be a potent GKAin vitro but failed to show detectable oral exposure in rat pharmacokinetic studies. This was hypothesised to be due to poor solubility linked to the high lipophilicity (logD) of the compound limiting absorption. Therefore, we believed that oral exposure might be improved if lipophilicity could be reduced. The challenge was to find a way to do this without losing potency. In the pyridine acid sub-series, SAR investigations of the biaryl ether substructure had revealed that potency could be maintained with polar groups such as the 4-methanesulfonyl derivative 3.12 However, these overall very polar compounds failed to show oral exposure, presumably now due to poor permeability. These two conflicting phenomena apparently limiting absorption and, hence, oral exposure in the series (poor solubility at high logD and poor permeability at low logD) are supported by in vitro measurements of solubility and Caco-2 flux (Table 1). It is possible that clearance, in addition to absorption, further impacts on oral exposure and indeed in vitro metabolic stability data (not shown) suggest that this may be the case at high logD
| GKA60 | 3 | 4 | |
|---|---|---|---|
|

|

|
|
| GK EC50/μM | 0.09 | 0.06 | 0.07 |
| LogD | 1.4 | -0.4 | 4.1 |
| Solubility/μM | 805 | >3000 | <0.7 |
| Caco-2 A to B Papp/nm s−1 | 74 | 3 | — |
| Rat oral nAUC/μmol.h.kg−1 | 8.2 | 0 | 0 |
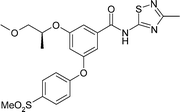
These observations allowed us to formulate a hypothesis that more polar neutral compounds targeting an intermediate logD range would achieve oral exposure and that incorporation of polar groups such as 4-methanesulfonyl to the pendant phenyl ring may provide a way to achieve such ends without compromising potency. In the event, although achieving the correct balance of lipophilicity appeared to be required to achieve oral exposure, it was not necessarily enough on its own and many compounds in the correct logD range still fail to achieve oral exposure. This is possibly due to high clearance although factors other than lipophilicity may also influence permeability and solubility However, we were gratified to find that compounds such as GKA70 and GKA71 achieve the necessary oral exposure with maintained potency such that they achieve activity in vivo as evidenced by their efficacy in a rat oral glucose tolerance test (OGTT)12 at 10 and 3 mgkg−1 respectively (Table 2). Thus, by testing GKA71, we were able to establish in rat studies that these compounds were free of the testicular toxicology observed with GKA50 and GKA60, proving that GK activation was not the cause of the toxicology and providing a way forward for the series.
| GKA70 | GKA71 | |
|---|---|---|

|
|
|
| GK EC50/μM | 0.06 | 0.11 |
| LogD | 3.2 | 2.9 |
| Solubility/μM | 20 | 129 |
| Caco-2 A to B Papp/nms−1 | 540 | 290 |
| hERG IC50/μM | 6.9 | 7.5 |
| Rat oral nAUC/μmol.h.kg−1 | 0.8 | 8.3 |
| Rat OGTT AUC reduction | 29% at 10 mgkg−1 | 27% at 3 mgkg−1 |
Whilst the cause of the testicular toxicology was not known at the time (and identifying its cause was not required in order to formulate a strategy to overcome the problem), we were aware of reports in the literature showing that pan-retinoic acid receptor (RAR) antagonists such as BMS-18945313 had shown similar effects. Once the basis for these effects had been determined, it was possible to link these observations together with some confidence. Comparing the structures of such compounds reveals a similarity between the benzoic acid containing portion of BMS-189453 (Fig. 3) and the pyridine acid of GKA50 and GKA60.
 | ||
| Fig. 3 BMS-189453. | ||
Subsequent testing of the pyridine acids exhibiting testicular toxicology (GKA50, GKA60 and compounds 1 and 2) revealed that they are all potent RAR-α antagonists (Table 3). Moreover, they exhibit, to a varying degree, diminished activity against the RAR-β and -γ isoforms, suggesting that RAR-α antagonism is the cause of the testicular toxicology exhibited by these compounds.
| Compound | GKA50 | GKA60 | 1 | 2 | GKA70 | GKA71 |
|---|---|---|---|---|---|---|
| RARα IC50/μM | 0.47 | <0.001 | 0.011 | 0.01 | >30 | >30 |
| RARβ IC50/μM | 32 | <0.001 | 0.007 | 0.135 | — | — |
| RARγ IC50/μM | >1000 | 19 | 1.2 | 22 | — | — |
| Testicular toxiciology in vivo | Yes | Yes | Yes | Yes | NA | No |
In support of this, genetic mutation studies of RAR-α in mice have also shown testicular toxicology.14 We therefore believe that in vitro screening against RAR-α antagonism may provide a useful means of avoiding such effects in future drug discovery programmes, in particular for compounds with structural similarity such as 4-substituted benzoic acids and heterocyclic analogues. GKA70 and GKA71 were found to be inactive towards RAR-α, consistent with this hypothesis. Of course this does not mean that antagonism of other isoforms of RAR would not cause testicular effects, indeed reports in the literature suggest that this may be the case, for example RAR-β antagonists have been shown to reduce agonist induced Sertoli cell proliferation.15
Protein crystal structures of RAR-α (PDB code: 3KMR) and glucokinase (PDB code: 3A0I) were used to post-rationalise the structural basis of these effects. The bound ligand of the glucokinase structure was first replaced with GKA60 using pharmacophore and shape similarity. The two protein structures were then overlaid on the basis of common ligand structural motifs, i.e. the benzoic or pyridine acid, the two atom linker and a second aromatic ring. The overlay shows that the size and shape of the binding site pocket is similar between the two proteins. On further examination we observe that the carboxylic acid moiety of the RAR-α antagonist makes a key interactions with the protein: Arg276 and the backbone of Ser287 (Fig. 4). These residues enclose one end of the binding pocket in RAR-α. Conversely the carboxylic acid was not observed to make any significant interactions with the GK protein, indeed the pocket is quite open to solvent at this location. It was our hypothesis that the carboxylic acid group remained partially solvated upon ligand binding to GK, conveying beneficial physical properties but not significantly effecting binding affinity. This would explain why it was possible to remove the carboxylate and maintain GK activity but with the need to rebalance the physical properties by introducing polar groups elsewhere in the molecule.
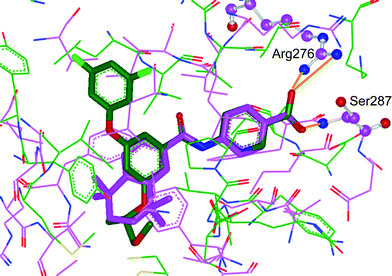 | ||
| Fig. 4 Overlaid crystal structures of glucokinase (PDB code: 3A0I) in green and RAR-α (PDB code: 3KMR) in purple. The ligand of 3A0I has been replaced with GKA60 on the basis of common pharmacophore elements. Protein–ligand interactions are marked in orange. | ||
Conclusions
In summary, we have shown that our previously disclosed lead compounds from the pyridine-5-carboxylic acid containing series of GKAs cause testicular toxicology, these effects can be attributed to antagonism of RAR-α and that non-acid containing compounds from the series do not possess the same liability. Moving from acidic to neutral compounds within the series requires a balancing of lipophilicity in order to maintain oral exposure and drug-like properties (Fig. 5). In the proceeding paper, we describe our efforts to further optimise these compounds to provide compounds suitable for clinical development. | ||
| Fig. 5 Balancing physical properties to achieve oral exposure with neutral compounds. | ||
Acknowledgements
This research was sponsored by AstraZeneca. All authors were AstraZeneca employees at the time of the conduct of the research. The authors thank the wider project team for synthesis and testing of the compounds and Dr Jeremy Burrows for helpful discussions that led to the identification of RAR as the target for the testicular toxicology.References
- M. Tadayyon and S. A. Smith, Expert Opin. Invest. Drugs, 2003, 12, 307 Search PubMed.
- R. C. Turner, C. A. Cull, V. Frighi and R. Holmann, J. Am. Med. Assoc., 1999, 281, 2005 Search PubMed.
- F. M. Matschinsky, Diabetes, 2002, 51, S394 Search PubMed.
- F. M. Matschinsky, M. A. Magnuson, in Frontiers in Diabetes, F. M. Matschinsky, M. A. Magnuson, ed. Karger:Basel, 2004;Vol. 16, pp 1–17 Search PubMed.
- A. Gloyn, S. Odili, D. Zelent, C. Buettger, H. A. Castleden, A. M. Steele, A. Stride, C. Shiota, M. A. Magnuson, R. Lorini, G. d'Annunzio, C. A. Stanley, J. Kwagh, E. van Schaftingen, M. Veigha da Cunha, F. Barbetti, P. Dunten, Y. Han, J. Grimsby, R. Taub, S. Ellard, A. T. Hattersley and F. M. Matschinsky, J. Biol. Chem., 2005, 280, 14105 Search PubMed.
- B. Leighton, A. Atkinson and M. P. Coghlan, Biochem. Soc. Trans., 2005, 33, 371 Search PubMed.
- D. Zelent, H. Najafi, S. Odili, C. Buettger, H. Weik-Collins, C. Li, N. Doliba, J. Grimsby and F. M. Matschinsky, Biochem. Soc. Trans., 2005, 33, 306 Search PubMed.
- For comprehensive reviews of GKAs see: M. C. T. Fyfe and M. P. Procter, Drugs Fut., 2009, 34, 641 Search PubMed; F. M. Matschinsky, Nat. Rev. Drug Discovery, 2009, 8, 399 Search PubMed; M. Coghlan and B. Leighton, Expert Opin. Invest. Drugs, 2008, 17, 145 CrossRef CAS.
- D. McKerrecher, J. V. Allen, S. Bowker, S. Boyd, P. W. R. Caulkett, G. S. Currie, C. D. Davies, M. L. Fenwick, H. Gaskin, E. Grange, R. B. Hargreaves, B. R. Hayter, R. James, K. M. Johnson, C. Johnstone, C. D. Jones, S. Lackie, J. W. Rayner and R. P. Walker, Bioorg. Med. Chem. Lett., 2005, 15, 2103 Search PubMed.
- D. McKerrecher, J. V. Allen, P. W. R. Caulkett, P. C. S. Donald, M. L. Fenwick, E. Grange, K. M. Johnson, C. Johnstone, C. D. Jones, J. W. Rayner and R. P. Walker, Bioorg. Med. Chem. Lett., 2006, 16, 2705 Search PubMed.
- K. G. Pike, J. V. Allen, P. W. R. Caulkett, D. S. Clarke, C. S. Donald, M. L. Fenwick, K. M. Johnson, C. Johnstone, D. McKerrecher, J. W. Rayner, R. P. Walker and I. Wilson, Bioorg. Med. Chem. Lett., 2011, 21, 3467 Search PubMed.
- G. Coope, A. M. Atkinson, C. Allott, D. McKerrecher, C. Johnstone, K. G. Pike, K. G. P. C Holme, H. Vertigan, D. Gill and M. P. Coghlan, Br. J. Pharmacol., 2006, 149, 328 Search PubMed.
- G. E. Schulze, R. J. Clay, L. E. Mezza, C. L. Bregman, R. A. Buroker and J. D. Frantz, Toxicol. Sci., 2001, 59, 297 Search PubMed.
- T. Lufkin, D. Lohnes, M. Mark, A. Dierich, P. Gorry, M.-P. Gaub, M. LeMeur and P. Chambon, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7225 Search PubMed.
- G. Livera, V. Rouiller-Fabre and R. Habert, Biol. Reprod., 2001, 64, 1307 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details for the synthesis of GKA71. See DOI: 10.1039/c1md00090j |
| ‡ Current address: Prosidion Limited, Watlington Road, Oxford, OX4 6LT, UK |
| This journal is © The Royal Society of Chemistry 2011 |