Blockage of collagen binding to integrin α2β1: structure–activity relationship of protein–protein interaction inhibitors†
Jarkko T.
Koivunen
ab,
Liisa
Nissinen
c,
Auni
Juhakoski
c,
Marjo
Pihlavisto
c,
Anne
Marjamäki
c,
Juhani
Huuskonen
bd and
Olli T.
Pentikäinen
*ab
aComputational Bioscience Laboratory, Department of Biological and Environmental Science, University of Jyväskylä, P.O.Box 35, FI-40014, Finland. E-mail: olli.t.pentikainen@jyu.fi; Fax: +358-14-260-2221; Tel: +358-40-521-6913
bNanoscience Center, University of Jyväskylä, Finland
cBiotie Therapies Corp., Turku, Finland
dDepartment of Chemistry, University of Jyväskylä, Finland
First published on 22nd June 2011
Abstract
The interaction between the α2β1 integrin and collagen plays a crucial role in the development of pathological conditions, such as thrombus formation and cancer cell metastasis. Accordingly, the α2β1 integrin is a promising target for the development of new drug molecules to treat these diseases. Here, we have designed, synthesized, and measured in vitro a set of novel drug-like compounds that block the protein–protein interactions between α2β1 integrin and collagen. The obtained structure–activity relationship reveals the key features that are required for successful inhibition of this integrin–collagen interaction.
Introduction
The integrins mediate bidirectional signaling between the cell and the extracellular matrix (ECM). The extracellular segment of the α2β1 integrin interacts with different types of collagens in the ECM, especially with collagen type I.1 In platelets, collagen binding to the α2β1 integrin on activates the outside-in signaling cascade and plays an important role in thrombus formation.2–4 Unlike some integrins, the collagen binding integrins do not recognize the peptide sequence Arg–Gly–Asp (RGD). Instead, several motifs from collagen type I (e.g. GFOGER)5 and collagen type III (e.g. GROGER and GLOGEN)6 are recognized by α2β1 via inserted domain in the α subunit (αI domain). Based on structural analyses, collagen binds to the αI domain of the α2β1 integrin, causing significant conformational change.7,8 Recent studies have also indicated that the interaction between the α2β1 integrin and collagen may have a crucial role in the development of thrombus9 and cancer metastasis.10 For example, prostate cancer metastasis has been shown to be mediated by the α2β1 integrin–type I collagen interaction.11 Studies have also indicated that integrin antagonists may have potentially therapeutic applications in the prevention of bone metastasis associated with prostate cancer.12 Other studies have proposed that the α2β1 integrin–collagen interaction may be a therapeutic target for the treatment of pancreatic cancer,13 as well as rhabdomyosarcoma.14Competitive blockage of protein–protein interactions, such as the α2β1 integrin–collagen interaction, is typically difficult to achieve even at the micromolar range.15 The binding affinity of the protein–protein complex dictates the limitations of the binding affinity of the small molecule. In the case of the α2β1 integrin–collagen interaction, a Kd value of 7.8 μM has been assigned to the triple helical mimetic peptide of collagen.16 The first peptide modulators that were derived from the snake venom of the Brazilian viper (Bothrops jararaca)17,18 have a similar Kd as the integrin αI domain of collagen peptide and an IC50 value of 1.2 μM.16 Previous study identified the binding sites of these peptides and competitive antagonists19 (Fig. 1: L3008; 1), and subsequently they were used in the development of a sulfonamide compound with antithrombotic activity20 (Fig. 1: BTT-3016; 2). 1 has an IC50 value of 7.5–12 μM at high concentrations. 1 is shown to be toxic, thus, the EC50 has not been determined.16,172, a compound that was used as a starting point in this study, was de novo designed into α2I domain by using visualization, and the binding site was verified with the single point mutation Tyr285Phe that did not demolish the collagen binding, but abolished the binding of 2 at the EC50 concentration.20 Numerous allosteric modulator molecules have also been developed21–23 such as the prolyl-2,3-diaminopropionic acid derivatives (DAPs in Fig. 1; 3). Compounds similar to 3 likely bind to the I-like domain of the β1-subunit,23 which is known to be a target for structurally similar compounds with other integrin α-subunits.24,25
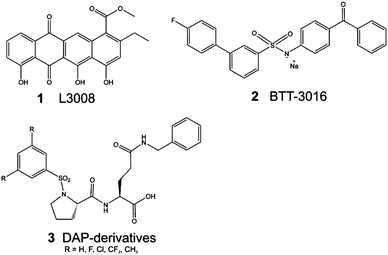 | ||
| Fig. 1 Previously published α2β1 integrin–collagen interaction modulators contain both allosteric, e.g.3,23 and competitive functioning antagonist ligands (116 and 217). | ||
Recent studies suggest that it is possible to develop small-molecule ligands that competitively block collagen binding to blood plateletsvia the α2β1 integrin.19,20 Elucidating the nature of the α2β1 integrin–collagen interaction could shed light on the importance of this integrin subunit for human health. With this aim in mind, we have here accomplished the first SAR study of α2β1 integrin competitive antagonist ligands, based on molecules that we have identified by employing rational target based drug design.19,20 The earlier described compounds, 1 and 2, were designed to compete with the binding of the triple helical collagen mimetic peptide that coordinates to the Mg2+ ion via the carboxylate group of glutamate.4 Accordingly, although the Mg2+ coordination is not optimally achieved with a sulfonamide compound, the combination of metal coordination with optimal shape of the compound enables successful discovery of competitive ligands for collagen binding into α2β1 integrin.
In this study we utilized a scaffold compound that we identified earlier20 (Fig. 1: 2) to develop a novel set of molecules. Despite the efficiency of 2 (EC50 = 2 μM) the high lipophilicity (log P: >5)20 sets limitations for its usage in drug development process. Thus, aim was to develop molecules with better solubility though some potency might be lost. Analysis of the structure–activity relationships of these α2β1 integrin antagonist ligands indicated that fairly small ligands (Mw ≈ 400 g mol−1) can block the binding of collagen to the α2β1 integrin. Therefore, our findings demonstrate that it is possible to rationalize the discovery of drug-like protein–protein interaction modulators, in this case between the αI-domain of α2β1 integrin and collagen.
Results and discussion
Molecular modeling
The 3D structure of the αI domain of the α2β1 integrin in its closed conformation state (pdb-code: 1aox8) was used as a target in the protein structure-based ligand discovery. Drawing on previous studies,19,20 we defined plausible pharmacophore points for efficient binding to the metal ion-dependent adhesion site of the α2β1 integrin (Fig. 2A): (1) coordination with Mg2+, (2) favorable hydrophobic contacts with the side chains of Leu286 and Leu291, (3) a hydrogen bond with the hydroxyl group of Tyr285, (4) a hydrogen bond with the main chain NH of Glu256, and (5) a water-mediated hydrogen bond with the main chain oxygen of Asp219. The significance of each pharmacophore point was explored using a series of molecules in which the structures of the ligands were changed one step at a time. To investigate whether the designed molecules satisfied the criteria set by the pharmacophore, docking studies of their binding capabilities were performed. | ||
| Fig. 2 Successful competitive antagonism of α2β1 integrin–collagen interaction requires both high shape complementarity and strong interactions between the integrin surface and the modulator molecule. (A) Pharmacophore for the ligand discovery derived from the binding site (1) Mg2+ (2) Leu286 and Leu291 (3) Tyr285 (4) Glu256 (5) Asp219. To fulfill the pharmacophore, a novel set of compounds (B) was developed. | ||
Based on de novo ligand discovery by using visualization the molecules were built up atom-by-atom, 2,4-dichlorobenzenesulfonyl was found to be a suitable sulfonyl moiety to replace the biphenylsulfonyl fragment of the previously discovered sulfonamide20 (Fig. 2B: yellow and Table 1). Furthermore, novel ligands based on 2,4-dichlorobenzenesulfonyl fragment have considerably lower log P values compared to the compound 2 (4: 3.5; 2: >5). Docking of 4 (Fig. 3A) suggested that one of the SO2 oxygens could facilitate the coordination to Mg2+. In addition, several other criteria of the pharmacophore (Fig. 2A) were fulfilled: the phenyl ring of the benzophenone fragment exerted hydrophobic effects on Leu286 and Leu291, the keto-oxygen formed a hydrogen bond with the main chain NH of Glu256, and the NH group formed a water-mediated (Fig. 3A–C: Wat3) hydrogen bond with the main chain oxygen atom of Asp219 (Fig. 3A and Table 1). Docking predicted that chloro-substituent at ortho position can form halogen bond26 with main chain oxygen of Asp219 (Fig. 3A and B). Additionally, ortho-chloro substituent could interact with hydrogen of water 3 (Fig. 3A and B). In addition, modeling suggested that the SO2 of 4 accepts a hydrogen bond from a water molecule (Fig. 3A: Wat1) that is coordinated with the Mg2+ ion. The EC50 value of 4 (EC50: 20 μM) indicates that 4 is a potent antagonist for α2β1 integrin.
| ID | Compound | Modelingb | Experimental results | |||
|---|---|---|---|---|---|---|
| Mg2+ Leu286/291 GLu256/Tyr285 Asp219 | EC50c/μM | Selectivityd (α2 EC50/α1EC50) | Inhibitione (%) 50 μM ± SEM Imax | Cyto-toxicityf | ||
| a Ni: no inhibition; Nd: not determined. b Modeling results obtained from the docking simulations: + indicates that the interaction with that group is present, note that in the case of Asp219 +w indicates that the interaction is water mediated. c EC50 (concentration required for half maximal effect) in CHO-α2 adhesion to collagen I. d Integrin α2 vs. α1 selectivity comparing EC50 values from CHO-α1 adhesion to collagen IV and CHO-α2 to collagen I (fold). e Inhibition % at 50 μM ± SEM (Standard error of measurement) and Imax (maximal inhibitory effect) at 100 μM in CHO-α2 adhesion to collagen I. f Cytotoxic effect on CHO wild type cells (−, not toxic). EC50 and inhibition measurements were repeated 2–4 times as triplicate for each compound. | ||||||
| 4 |
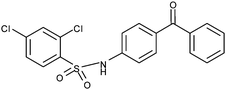
|
+ | 20 | 1.5 | 82 ± 1, 82 | (−) ≤20 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 5 |

|
+ | 17 | 3.0 | 69 ± 4, 69 | (−) ≤200 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 6 |
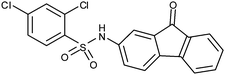
|
+ | 20 | 1.2 | 93 ± 1, 93 | (−) ≤200 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 7 |

|
+ | Ni | Nd | 0, Nd | Nd |
| − | ||||||
| + | ||||||
| +w | ||||||
| 8 |

|
−(+) | Ni | Nd | 0, Nd | Nd |
| − | ||||||
| −(+) | ||||||
| − | ||||||
| 9 |

|
− | Ni | Nd | 8, Nd | Nd |
| − | ||||||
| − | ||||||
| − | ||||||
| 10 |
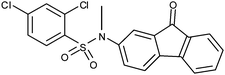
|
− | Ni | Nd | 0, Nd | Nd |
| − | ||||||
| − | ||||||
| − | ||||||
| 11 |

|
−(+) | Ni | Nd | 0, Nd | Nd |
| − | ||||||
| −(+) | ||||||
| − | ||||||
| 12 |

|
+ | 19 | 0.8 | 70 ± 19, 87 | (−) ≤200 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 13 |
|
− | Ni | Nd | 4, Nd | (−) ≤200 μM |
| − | ||||||
| − | ||||||
| − | ||||||
| 14 |

|
− | Ni | Nd | 0, Nd | (−) ≤200 μM |
| − | ||||||
| − | ||||||
| − | ||||||
| 15 |

|
− | Ni | Nd | 35 ± 5, 35 | (−) ≤200 μM |
| − | ||||||
| − | ||||||
| − | ||||||
| 16 |

|
− | Ni | Nd | 34 ± 9, 34 | (−) ≤200 μM |
| − | ||||||
| − | ||||||
| − | ||||||
| 17 |

|
+ | 16 | 2.2 | 92 ± 1, 96 | (−) ≤200 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 18 |
|
− | Ni | Nd | 34 ± 4, 34 | (−) ≤200 μM |
| − | ||||||
| − | ||||||
| − | ||||||
| 19 |
|
+ | 22 | 1.3 | 85 ± 7, 85 | (−) ≤200 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 20 |

|
+ | 40 | Nd | 72 ± 15, 72 | (−) ≤200 μM |
| + | ||||||
| + | ||||||
| +w | ||||||
| 21 |

|
+ | 11 | 2.8 | 86 ± 4, 95 | (−) ≤100 μM |
| + | ||||||
| + | ||||||
| + | ||||||
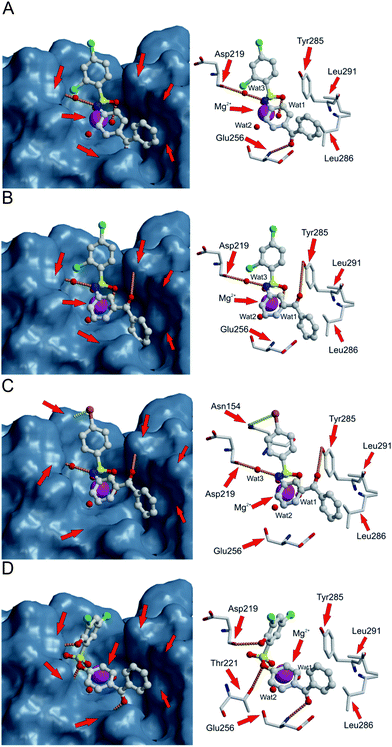 | ||
| Fig. 3 Compounds 4 (A), 5 (B), 17 (C) and 21 (D) docked into the collagen binding site of the αI domain of the α2β1 integrin. The hydrogen bonds are shown with orange dotted lines, favorable interaction between the bromine and NH-group is shown with yellow dotted line, hydrogen atoms are omitted for clarity. To illustrate the shape complementarity between the bound ligand and the αI domain, the surface of the αI domain is shown in the left panel, while the right panel shows in more detail the interactions that take place. | ||
The effect of structural isomerism was explored by changing the 4-aminobenzophenone moiety of 4 to 3-aminobenzophenone (compound 5; Fig. 3B and Table 1). Docking suggested that 5 could form very similar interactions with the integrin as 4. It coordinated with Mg2+viaSO2 and formed a hydrogen bond with Mg2+-coordinated water (Fig. 3B: Wat1). The NH group formed a water-mediated hydrogen bond with the main chain oxygen of Asp219 (Fig. 3B, Wat3), the chloro in the ortho position was oriented in the similar way as with 4, and the phenyl ring of the benzophenone fragment was able to make hydrophobic contact with the side chains of Leu286 and Leu291. In addition, modeling indicated that 5 could form a hydrogen bond with the hydroxyl group of Tyr285 (Fig. 3B), in addition to hydrogen bond with the main chain NH of Glu256 (4; Fig. 3A and Table 1). However, this hydrogen bond is not geometrically optimal and the distance is rather long (3.3 Å). Based on the docking results, 5 has optimal shape as it fills the binding pocket effectively. The packing of benzophenone into the bottom of the ligand-binding cavity between Glu256 and Leu286 is also more efficient in 5 than in 4.
In both 4 and 5, the aromatic rings of benzophenone favor a perpendicular orientation to each other. However, as previously shown with tetracycline 119 (Fig. 1), the cavity lined by Glu256 and Leu286 and Leu291 is very narrow and could, thus, favor a more planar ligand. Accordingly, benzophenone was replaced with 9-fluorenone, a planar fragment, in molecule 6 (Table 1). Docking studies suggested that 6 could form similar interactions with the αI domain of the α2β1 integrin as 4 (Table 1). In addition, as 6 is a more rigid ligand than 4, the 9-fluorenone moiety of 6 adheres more tightly against the hydrophobic side chains of the amino acids Leu286 and Leu291 than the benzophenone of 4. In vitro measurements showed that the EC50 values of these ligands are quite similar (4: 20 μM, 5: 17 μM, and 6: 20 μM; Table 1). However, analysis of the maximal inhibition of all of the molecules revealed that 6 is slightly more efficient than 4 and 5 (Table 1).
To explore the importance of an additional pharmacophore criterion—hydrophobic contacts with the side chains of Leu286 and Leu291—the benzophenone fragment was replaced with a significantly smaller acetophenone fragment (7; Table 1). The docking of 7 produced a highly similar conformation to that of 4 (data not shown), but the pharmacophore criterion was not fulfilled. In vitro, 7 exhibited no antagonist activity; consequently, hydrophobic contacts with Leu286 and Leu291 seem to play an important role in successful antagonism of collagen binding to the αI domain of the α2β1 integrin. Published crystal structures of the “closed” (pdb-code: 1aox8) and “open” (pdb-code: 1dzi7) αI domains of the α2β1 integrin have shown that the region containing Leu286 and Leu291 undergoes a significant conformational change upon collagen binding. It is, therefore, reasonable to speculate that the acetophenone in this region is unable to stabilize the αI domain structure efficiently enough to combat the conformational change induced by collagen binding. Taken together, the modeling and biological analyses imply that altering this region (Fig. 2B: green) so that it contains 3-substituted benzophenone rather than 4-substituted or so that it has a more rigid 9-fluorenone has only minor consequences for the antagonist activity of 2,4-dichlorosulfonamide derivatives. They also suggest that a smaller substituent (7) fails to inhibit collagen binding to the αI domain of the α2β1 integrin.
As noted above for 4 and 5 (Fig. 3A and B), modeling suggested that the NH in the sulfonamide moiety donates a hydrogen bond to the water molecule (Fig. 3: Wat3), which donates a hydrogen bond to the main chain oxygen atom of Asp219. This interaction, and, more generally the importance of donor function of the sulfonamide moiety (Fig. 2B: red) was explored with two modifications: (A) based on modeling, N-methylation would be expected to displace the water molecule (Fig. 3: Wat3), and the N-methyl would form an unfavourable interaction with the main chain oxygen atom of Asp219. This problem was clearly shown by the ligand docking results of these compounds, with Mg2+ coordination occurring viaketo-oxygen (rather than SO2) and SO2 forming a hydrogen bond with the main chain NH of Glu256 (data not shown). (B) Similarly, when sulfonamide was replaced with sulfonic ester (11), the modeling indicated that the water molecule is between the two hydrogen bond acceptors—ester oxygen of 11 and the main chain oxygen atom of Asp219. Thus, this water molecule should simultaneously donate hydrogen bonds in two directions that are exactly opposite that of the water molecule, which is impossible. In common with 8, 11 adopted a docking conformation where Mg2+ coordination was formed through keto-oxygen, rather than SO2 (data not shown). The in vitro results confirmed that all four compounds lacking hydrogen bond donor function (8–11) are functionally inactive (Table 1); thus, the hydrogen bond donor plays a significant role in this set of molecules.
The docking results suggested that the keto-oxygen of molecules 4–6 can form a hydrogen bond with the main chain NH of Glu256 or the side chain OH of Tyr285. The replacement of fluorenone (6) with fluorene (12) maintained the shape of the molecule and enabled us to explore the significance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group for α2β1 integrin antagonism. Surprisingly, the fluorene derivative (12) docked to the binding site in a very similar manner to that of 6, although it lacked one hydrogen bond acceptor and, as a consequence, a possible hydrogen bond. As a result, 12 could be expected to perform less efficiently than 6 in terms of blocking the collagen binding. However, the in vitro measurements showed that the EC50 values are practically the same for both compounds (12: 19 μM; 6: 20 μM; Table 1). This finding may be explained by a number of factors. (1) The removal of oxygen may increase the lipophilicity of the molecule; thus, binding of 12 (log P: 4.1) would be more entropy-driven than that of 6 (log P: 3.4). (2) The CH2-group of the fluorene exhibits a slight positive charge, enabling it to interact favorably with the hydroxyl-group of Tyr285. (3) The main chain angles between Gly255 and Glu256 can undergo conformational change, as seen in the comparison of the closed8 and open7 conformations of the αI domain of the α2β1 integrin (data not shown); as a result, fluorene can pack favourable against this conformationally altered region.
O group for α2β1 integrin antagonism. Surprisingly, the fluorene derivative (12) docked to the binding site in a very similar manner to that of 6, although it lacked one hydrogen bond acceptor and, as a consequence, a possible hydrogen bond. As a result, 12 could be expected to perform less efficiently than 6 in terms of blocking the collagen binding. However, the in vitro measurements showed that the EC50 values are practically the same for both compounds (12: 19 μM; 6: 20 μM; Table 1). This finding may be explained by a number of factors. (1) The removal of oxygen may increase the lipophilicity of the molecule; thus, binding of 12 (log P: 4.1) would be more entropy-driven than that of 6 (log P: 3.4). (2) The CH2-group of the fluorene exhibits a slight positive charge, enabling it to interact favorably with the hydroxyl-group of Tyr285. (3) The main chain angles between Gly255 and Glu256 can undergo conformational change, as seen in the comparison of the closed8 and open7 conformations of the αI domain of the α2β1 integrin (data not shown); as a result, fluorene can pack favourable against this conformationally altered region.
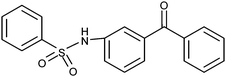
Next, in addition to varying the amino end of the molecule (4–12; Fig. 2B: green), we explored the effect of changes at the sulfonyl site (Fig. 2B: yellow). The amino end was still systematically varied with both 3- and 4-aminobenzophenones (Fig. 2B: green). The used pharmacophore (Fig. 2A) does not contain features at the sulfonyl site (Fig. 2A and B: yellow). First, both chlorides were removed, resulting in compounds 13 and 14. Neither of these compounds antagonized the α2β1 integrin–collagen interaction (Table 1). In contrast, the 4-methylbenzene derivatives (15 and 16) exhibit weak α2β1 integrin antagonism as both 3- and 4-benzophenone derivatives (maximum inhibition 35% and 34% for 15 and 16, respectively; Table 1).
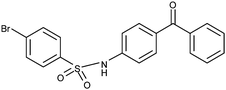
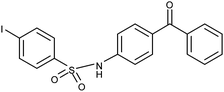
Compounds 17 and 18 contain bromide at the 4-position. Compared with the di-chloride compounds 4 and 5, both 4-bromobenzene derivatives would be expected to exhibit similar activity. However, the 3-benzophenone derivative (17) is highly potent antagonist (Table 1: EC50: 16 μM; 92% inhibition at 50 μM concentration; and maximal inhibition 96%), while compound 18 showed only weak inhibition at 50 μM concentration (34%; Table 1). In addition, 17 has low log P value (3.1). The surprisingly good antagonist activity of 17 may be attributable to the slight shift of the phenyl-ring orientation, in contrast to 4 and 5, resulting in a favorable interaction between the bromine of 17 and the side chain carbonyl oxygen of Asn154 (Fig. 3C), while 18 is forced to point the bromine towards the solvent rather than Asn154. Thus, interactions formed by halogen substituents of the sulfonyl moiety appear to be important. In contrast to the bromo-derivatives, the compound pair 19 and 20 with 4-iodobenzene at the sulfonyl end of the molecule showed the opposite trend, with the 4-aminobenzophenone derivative 19 being a 2-fold better antagonist (EC50: 22 μM) than the 3-aminobenzophenone derivative 20 (EC50: 40 μM; Table 1). Docking of these molecules to the αI domain of the α2β1 integrin indicated that large iodo-substituent makes 20 slightly too bulky to fit well into the binding cavity, and in addition, the interaction with the Asn154 is not as favorable as with bromine of 17. Also, the docking of 19 was not sterically hindered as much as that of 20 (data not shown), thereby explaining why 19 is a better antagonist than 20.
To further validate the proposed pharmacophore, we explored the possibility to replace the water mediated interaction of ligands with the main chain oxygen atom of Asp219 to the direct hydrogen bonding interaction. The resulting molecule (21) fulfills all set criteria to the successful binding, although the orientation of the phenyl ring at the sulfonyl end of the molecule does not remain optimal in terms of stacking with Tyr285 (Fig. 3D and Table 1). However, the slight rotation of the phenyl ring allows the 2-hydroxyl to form a hydrogen bond with the main chain oxygen atom of the Asp219. Furthermore, the change in the orientation of the phenyl also reflects into central core position, and accordingly, the ester oxygen can accept a hydrogen bond from the hydroxyl group of Thr221. This hydrogen bond is not seen with bound sulfonamides (Fig. 3). It is notable that the amino end of the molecule can still accept a hydrogen bond from the main chain NH of Glu256. The favorability of the binding can be also seen from the biological activity: EC50 (11 μM) visualizes that 21 is nearly twice as potent as 4. Moreover, the log P value of 21 (3.1) is lower than that of 4 (3.5). Although 21 has instead of sulfonamide, a sulfonyl ester that was found to make compound 11 inactive, the addition of 2-hydroxyl group successfully replaces the required donor function of the NH group.
The αI domains of the collagen binding integrins are highly conserved, making it difficult to achieve ligand selectivity between different subtypes (Fig. 4).17,27 All of the compounds were tested to explore their selectivity between the αI domains of α2β1 and α1β1 integrins. This was done because α1I domain closely resembles that of α2, and is the key determinant for the subtype selectivity, while other collagen-binding integrins with β1 subunit have not shown activity for sulfonamides.20 At the pharmacophore site, the αI domain ligand binding site of the α128 integrin differs from α28 integrin in two positions, with Tyr285 and Leu286 in α2 being replaced by Ser284 and Tyr285 in α1, respectively (Fig. 4) Although some ligands show minor selectivity to α2 over α1 no significant selectivity was achieved. However, tested molecules can give some idea about selectivity between α2 and α1. For example, active compounds containing keto-oxygen (4, 6, 17–20) exhibited slight α2 selectivity over α1 (Table 1). These results imply that the acceptor function of keto-oxygen may have role in the creation of the α2 selective ligand. In addition, the ligands based on the 3-aminobenzophenone fragment (5, 17, 20) tend to be slightly more selective to α2 than α1 compared with those based on 4-aminobenzophenone (4, 18, 19; Table 1).
 | ||
| Fig. 4 Subtype selective ligand-binding between α2β1 and α1β1 integrin can be achieved by targeting the ligands into αI domain. Superimposed αI domains of α2β1 (pdb-code: 1aox;8 blue cartoon representation) and α1β1 integrin (pdb-code: 1pt6;28 green cartoon representation) with the key differences at the binding site: Tyr285-Leu286 of α2 (white carbon atoms) is replaced in α1 by Ser284-Tyr285 (black carbon atoms). | ||
Conclusions
A set of novel α2β1 integrin ligands were designed, synthesized, and measured in vitro to verify the key properties of integrin antagonism. Analysis of the structure–activity relationship of this molecular series verified the following: (1) the model used is valid for the rational discovery of small molecules that can inhibit the adhesion of α2β1 integrin to collagen. (2) The NH donor of sulfonamide is critical for successful α2β1 binding. (3) The structural isomerism of benzophenone has no significant effect on the binding potency of α2β1, but it seems to have an impact on the selectivity of the integrin subtype. (4) The keto-oxygen of the amino moiety has a role in the selectivity of α2 compared to α1. (5) The halogen substituent is important for efficient ligand binding. Our results pinpoint the main features of small, drug-like sulfonamide molecules that are able to block the binding of collagen to the α2β1 integrin. Furthermore, we were able to develop biologically molecules having significantly lower log P values compared to ligands discovered earlier. Together, these results provide a firm foundation for the development of novel α2β1 integrin-mediated pharmaceutical agents for use in the prevention of thrombus formation and cancer cell metastasis.References
- M. M. Zutter and S. A. Santoro, Am. J. Pathol., 1990, 137, 113 Search PubMed.
- S. Niewiarowski, R. K. Stuart and D. P. Thomas, Proc. Soc. Exp. Biol. Med., 1966, 123, 196 Search PubMed.
- R. W. Farndale, J. J. Sixma, M. J. Barnes and P. G. de Groot, J. Thromb. Haemostasis, 2004, 2, 561 Search PubMed.
- A. B. Herr and R. W. Farndale, J. Biol. Chem., 2009, 284, 19781 CrossRef CAS.
- C. G. Knight, L. F. Morton, A. R. Peachey, D. S. Tuckwell and R. W. Farndale, et al., J. Biol. Chem., 2000, 275, 35 CrossRef CAS.
- N. Raynal, S. W. Hamaia, P. R. Siljander, B. Maddox and A. R. Peachey, et al., J. Biol. Chem., 2006, 281, 3821 CrossRef CAS.
- J. Emsley, C. G. Knight, R. W. Farndale, M. J. Barnes and R. C. Liddington, Cell, 2000, 101, 47 CrossRef CAS.
- J. Emsley, S. L. King, J. M. Bergelson and R. C. Liddington, J. Biol. Chem., 1997, 272, 28512 Search PubMed.
- S. M. Sweeney, G. DiLullo, S. J. Slater, J. Martinez and R. V. Iozzo, et al., J. Biol. Chem., 2003, 278, 30516 CrossRef CAS.
- W. Guo and F. G. Giancotti, Nat. Rev. Mol. Cell Biol., 2004, 5, 816 CrossRef CAS.
- C. L. Hall, J. Dai, K. L. van Golen, E. T. Keller and M. W. Long, Cancer Res., 2006, 66, 8648 Search PubMed.
- H. L. Goel, J. Li, S. Kogan and L. R. Languino, Endocr. Relat. Cancer, 2008, 15, 657 Search PubMed.
- J. J. Grzesiak and M. Bouvet, Br. J. Cancer, 2006, 94, 1311 Search PubMed.
- B. M. Chan, N. Matsuura, Y. Takada, B. R. Zetter and M. E. Hemler, Science, 1991, 251, 1600 Search PubMed.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301 CrossRef CAS.
- L. J. Lambert, A. A. Bobkov, J. W. Smith and F. M. Marassi, J. Biol. Chem., 2008, 283, 16665 Search PubMed.
- O. Pentikäinen, A. M. Hoffrén, J. Ivaska, J. Käpylä and T. Nyrönen, et al., J. Biol. Chem., 1999, 274, 31493 Search PubMed.
- J. Ivaska, J. Käpylä, O. Pentikäinen, A. M. Hoffrén and J. Hermonen, et al., J. Biol. Chem., 1999, 274, 3513 Search PubMed.
- J. Käpylä, O. T. Pentikäinen, T. Nyrönen, L. Nissinen and S. Lassander, et al., J. Med. Chem., 2007, 50, 2742 Search PubMed.
- L. Nissinen, O. T. Pentikäinen, A. Jouppila, J. Käpylä and M. Ojala, et al., Thromb. Haemostasis, 2010, 103, 387 Search PubMed.
- H. Yin, L. O. Gerlach, M. W. Miller, D. T. Moore and D. Liu, et al., Bioorg. Med. Chem. Lett., 2006, 16, 3380 CrossRef CAS.
- S. Choi, G. Vilaire, C. Marcinkiewicz, J. D. Winkler and J. S. Bennett, et al., J. Med. Chem., 2007, 50, 5457 Search PubMed.
- M. W. Miller, S. Basra, D. W. Kulp, P. C. Billings and S. Choi, et al., Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 719 Search PubMed.
- D. Heckmann, B. Laufer, L. Marinelli, V. Limongelli and E. Novellino, et al., Angew. Chem., Int. Ed., 2009, 48, 4436 Search PubMed.
- D. Heckmann, A. Meyer, L. Marinelli, G. Zahn and R. Stragies, et al., Angew. Chem., Int. Ed., 2007, 46, 3571 Search PubMed.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061 CrossRef CAS.
- M. Tulla, O. T. Pentikäinen, T. Viitasalo, J. Käpylä and U. Impola, et al., J. Biol. Chem., 2001, 276, 48206 Search PubMed.
- Y. Nymalm, J. S. Puranen, T. K. M. Nyholm, J. Käpylä and H. Kidron, et al., J. Biol. Chem., 2004, 279, 7962 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting quantum mechanics and adhesion data; materials and methods, including experimental and spectroscopic details for the synthesized compounds. See DOI: 10.1039/c1md00089f |
| This journal is © The Royal Society of Chemistry 2011 |