HIV-1 protease inhibitors with a tertiary alcohol containing transition-state mimic and various P2 and P1′ substituents†
Per
Öhrngren
a,
Xiongyu
Wu
a,
Magnus
Persson
b,
Jenny K.
Ekegren
a,
Hans
Wallberg
c,
Lotta
Vrang
c,
Åsa
Rosenquist
c,
Bertil
Samuelsson
c,
Torsten
Unge
b and
Mats
Larhed
*a
aDepartment of Medicinal Chemistry, Organic Pharmaceutical Chemistry, BMC, Uppsala University, Box 574, SE-751 23, Uppsala, Sweden. E-mail: mats.larhed@orgfarm.uu.se; Fax: +46 18 4714374; Tel: +46 18 4714667
bDepartment of Cell and Molecular Biology, Structural Biology, BMC, Uppsala University, Box 596, SE-751 24, Uppsala, Sweden
cMedivir AB, PO Box 1086, SE-141 22, Huddinge, Sweden
First published on 26th May 2011
Abstract
Two series, including in total 18 novel HIV-1 protease inhibitors, comprising a tertiary alcohol as the transition-state mimic have been synthesised and evaluated. Replacement of the previously used, but metabolically unstable, indanol amide group with amino acid derived aliphatic P2–P3 moieties provided potent inhibitors with low Ki- and EC50-values (2.7 nM and 2.0 μM, respectively). The P1′ subunit was varied using 10 different aromatic and heteroaromatic substituents furnishing the corresponding inhibitors with retained activity. Permeability and stability studies showed examples in the same range as Atazanavir. X-Ray crystallographic analysis of two selected inhibitor enzyme co-complexes (9a and 9d) supplied detailed structural information. The binding modes were compared to those of Atazanavir and a previously reported indanol amide containing inhibitor (14). The novel inhibitors with an elongated P1′ side chain enabled a previously unexploited edge-on interaction with Phe53/153. Exchange of the previously used indanol amide P2 moiety, with a tert-leucine derived P2–P3 side chain, furnished small main chain displacements in the S2–S3 pocket. The methyl amide in the P3 position caused a 2 Å shift of the Arg8/108 in comparison to 14, indicating the flexibility of the protease active site.
Introduction
As constituents of the highly active antiretroviral therapy (HAART), the HIV-1 protease inhibitors represent a landmark in the treatment of HIV/AIDS. By reducing the viral loads, a higher quality of life and also a prolonged life-expectancy have been achieved in patients using this therapy.1–5 However, low patient compliance, severe adverse effects and a rapidly increasing number of resistant viral strains contribute to the need for new, structurally unique entities to be evaluated for anti-HIV activity.5–12 The design, synthesis and evaluation of novel HIV-1 inhibitors to efficiently meet these needs are therefore highly desirable.We have previously reported a new type of transition-state mimic designed for use in HIV-1 protease inhibitors: a tertiary alcohol, which in the proper structural environment provides highly potent compounds, as exemplified by compound A (Fig. 1).13–15 Further elaboration of the core structure of these inhibitors leads to compounds of classes B16 and C17 (Fig. 1).
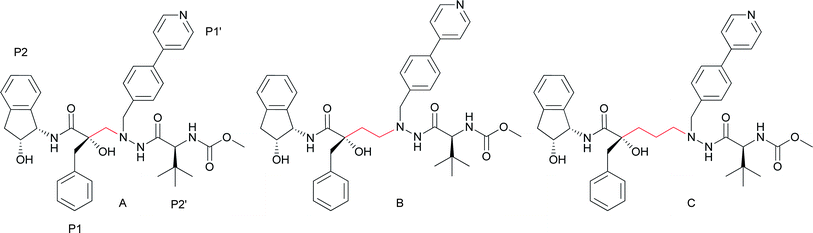 | ||
| Fig. 1 Examples from previously reported series of tertiary alcohol containing HIV-1 protease inhibitors: A with a one carbon backbone spacer (Ki = 5.5 nM),14B with a two carbon backbone spacer (Ki = 2.3 nM)16 and C with a three carbon backbone spacer (Ki = 2.8 nM).17 Spacers indicated in red. | ||
The P2 and P1′ groups18 in these types of inhibitors have been shown to be of particular interest. The (1S,2R)-1-amido-2-indanol group, used as the P2 substituent of inhibitors A–C, is rapidly oxidised by metabolic enzymes.19,20 In addition, the chemical nature of the P1′ side chain is known to strongly influence the antiviral activity of the compounds in cell-based assays.14,15,17,21
With this background, we were interested in developing a straightforward synthetic approach for the incorporation of new amino acid derived P2–P3 groups into compound C, since amino acid derivated P2–P3 substituents have been successful in the case of the approved inhibitor Atazanavir (Atz) (Fig. 2).21,22 Furthermore, we aimed at optimising the most potent derivative in the P2–P3 series by creating a small library of compounds with various aromatic P1′ substituents using microwave accelerated, palladium-catalysed Suzuki–Miyaura23,24 and Sonogashira25,26 reactions. Within this article we present the synthetic protocols and the inhibitory potency on enzyme and in cell based assay of the final products, together with X-ray evaluation of two of the inhibitors.
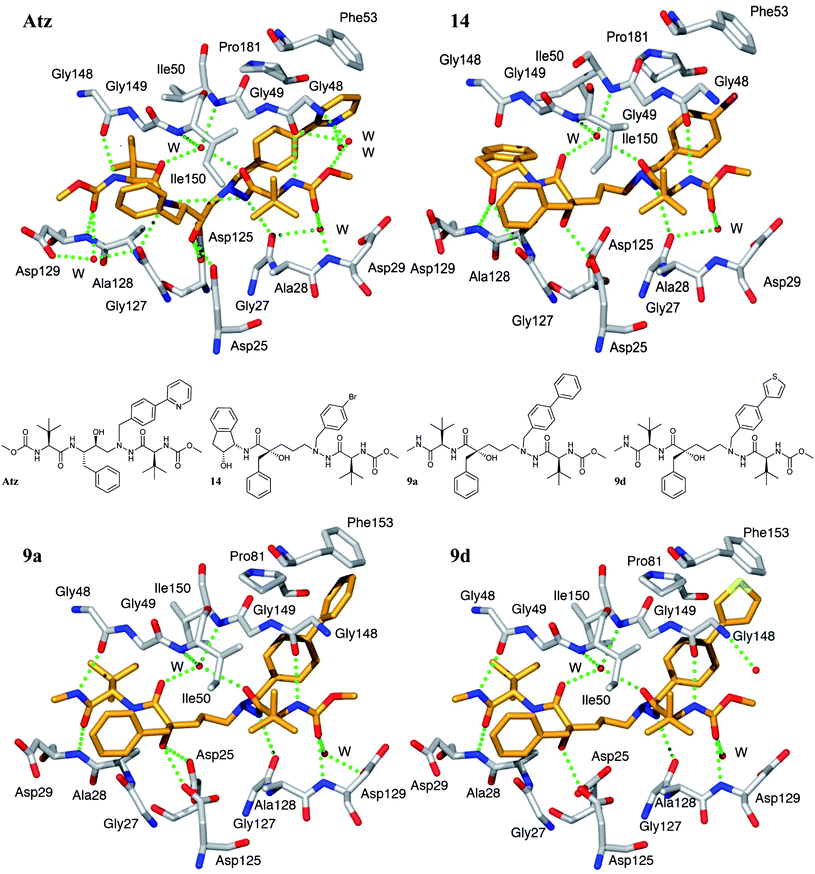 | ||
| Fig. 2 Comparison of the overall conformations and binding patterns of the compounds Atz (top left, PDB code 3EL9), 14 (top right, PDB code 2uxz), 9a (bottom left, PDB code 2xye) and 9d (bottom right, PDB code 2xyf) in the active site of HIV-1 protease. The hydrogen bonds to the catalytic Asp25 and Asp125 are longer (∼3.3 Å) for 14, 9a and 9d, whereas Atz has two short hydrogen bonds (2.8 Å) and an additional one at 3.3 Å. Compounds 9a and 9d form 7 and 6 direct hydrogen bonds to the protease, respectively. Additionally, both compounds form 3 more hydrogen bonds viawater molecules, of which two are to the structural water. The corresponding numbers for 14/Atz are 5/9 direct hydrogen bonds and 3/5 viawater. Edge-on aromatic interactions with Phe53/153 are formed with the elongated P1′ side chains of 9a and 9d. | ||
Chemistry and results
Starting from compound 1, prepared according to a literature procedure,17 the P2–P3 varied inhibitors 7a–f were synthesised in four steps (Scheme 1). Hydrolysis of the dioxolane ring in 1 facilitated intramolecular γ-lactone formation between the free alcohol and the methyl ester unit resulting in acid 2. Peptide coupling of the acid and the different methyl-amide-derivatised amino acids (3a–e) or TBS protected L-tert-leucinol (3f), using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and 1-hydroxybenzotriazole (HOBt) at room temperature, gave compounds 4a–f (mixed diastereomers).27 The absolute configuration of the quaternary carbon in 4a–f was determined by X-ray crystallography of (S)-4a.28 Separation of the diastereomers by flash chromatography on silica was followed by a number of structural modifications in the (R)-derivatives, which, as deduced from previous results, was the preferred stereochemistry of the tertiary alcohol.13,15–17,29 | ||
| Scheme 1 Synthesis of inhibitors 7a–f encompassing different P2 substituents. Reagents and conditions: (a) TFA, H2O, 80 °C, 95%; (b) amino acid derivates 3a–f (for R-groups see Table 1), EDC, HOBt, DCM, room temp., 25–46% ((R)-4a–f), 47% ((S)-4a), diastereomers separated by flash chromatography; (c) (1) LiBH4, Et2O, room temp; (2) trimethylacetyl chloride, pyridine, room temp.; (3) TBSOTf, Et3N, DCM, 0 °C to room temp.; (4) LiBH4, Et2O, room temp., 25–62%; (d) (1) Dess–Martin periodinane, DCM, room temp.; (2) 6, Na(OAc)3BH, AcOH, THF, room temp.; (3) TBAF, THF, room temp., 3–81%. | ||
Next, reduction of the lactone moiety in 4, protection of the primary alcohol by pivaloyl chloride, protection of the tertiary alcohol with tert-butyldimethylsilyl triflate (TBSOTf) and finally, reduction of the pivaloyl ester with LiBH4 gave the alcohols 5a–f (Scheme 1). Compounds 5a–f were oxidised using Dess–Martin periodinane and thereafter, a reductive amination with hydrazide 6, synthesised as previously reported,15 was performed followed by deprotection of the TBS-group, which resulted in the target compounds 7a–f (Scheme 1).
The HIV-1 protease inhibition and the cell-based antiviral activity of 7a–f, as given by the Ki and the EC50 values, are summarised in Table 1 (inhibitor C17 is included as reference compound). Of inhibitors 7a–e, having lipophilic P2–P3 amino acidN-methyl amide side chains, only compounds 7a, c, d, carrying the shortest side chains, showed Ki values below 20 nM and EC50 values below 3 μM. Inhibitor 7a, with a tert-leucine derived P2-group, afforded one of the more potent compounds, measured on enzyme, and also the best cell-based antiviral activity in the series (Ki = 6.2 nM, EC50 = 2.0 μM). The inhibitors 7b and 7e, having the longest P2 side chains, showed the lowest activity with Ki-values above 50 nM. Inhibitor 7f was designed to conserve the hydrogen bond donating capacity of the hydroxyl group which was previously seen in inhibitors using the amino-indanol as P2–P3 fragment.14–17,30 However, this inhibitor furnished only low inhibition of the enzyme and did not show any activity in the cell based assay. Leaving the TBS-protection group on the tertiary alcohol was not beneficial, as demonstrated by inhibitor 8 which only afforded very low inhibition (Table 1). As expected, (S)-7a provided no HIV-1 protease inhibition.15
| Cmpd | R= | Yieldb (%) | K i c/nM | EC50d/μM | CC50/μM |
|---|---|---|---|---|---|
| a Conditions: see Scheme 1. b Isolated yields in the final reductive amination–deprotection step (d). c Atazanavir (Atz): Ki = 2.7 nM.33 d Atz: EC50 = 0.0039 μM.33 e P app (Caco-2) = 4.6 × 10−6 cm s−1, CLint = 180 μL min−1 mg−1. f S-Configuration at the tertiary alcohol. g 7a with the tertiary alcohol TBS-protected. | |||||
| C | — | — | 2.8 | 0.17 | — |
| 7a e |
|
66 | 6.2 | 2.0 | >10 |
| (S)-7af |
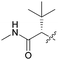
|
28 | >5000 | >10 | >10 |
| 7b |
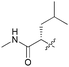
|
81 | 65 | >10 | >10 |
| 7c |
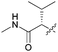
|
18 | 2.7 | 2.1 | >10 |
| 7d |
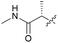
|
29 | 18 | 2.6 | >10 |
| 7e |
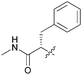
|
76 | 170 | 8.2 | >10 |
| 7f |
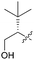
|
3 | 130 | >10 | >10 |
| 8 g |
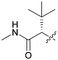
|
71 | 2700 | >10 | >10 |
Compound 7a was first chosen for further optimisation of the P1′ position, using sequential microwave-accelerated, palladium-catalyzed Suzuki–Miyaura cross-coupling reactions in sealed vessels (Scheme 2).31,32
![Microwave-accelerated synthesis of inhibitors 9a–h. Reagents and conditions: (1) Method A: Suzuki–Miyaura cross-coupling; 8, RB(OH)2, K2CO3, Herrmann's palladacycle, [(t-Bu)3PH]BF4, DME/H2O, 120 °C, 20 min, giving TBS-9a–e. Method B: Sonogashira cross-coupling; 8, ethynyl pyridine, PdCl2(PPh3)2, CuI, Et3N, DMF giving TBS-9g. Method C: copper free Sonogashira cross-coupling; 8, ethynyl pyridine, PdCl2(PPh3)2, piperidine, H2O/acetone, 130 °C, 60 min or 140 °C, 30 min, giving TBS-9f, h. (2) TBAF, THF, room temp., giving 9a–h in 14–82% yield.](/image/article/2011/MD/c1md00077b/c1md00077b-s2.gif) | ||
| Scheme 2 Microwave-accelerated synthesis of inhibitors 9a–h. Reagents and conditions: (1) Method A: Suzuki–Miyaura cross-coupling; 8, RB(OH)2, K2CO3, Herrmann's palladacycle, [(t-Bu)3PH]BF4, DME/H2O, 120 °C, 20 min, giving TBS-9a–e. Method B: Sonogashira cross-coupling; 8, ethynyl pyridine, PdCl2(PPh3)2, CuI, Et3N, DMF giving TBS-9g. Method C: copper free Sonogashira cross-coupling; 8, ethynyl pyridine, PdCl2(PPh3)2, piperidine, H2O/acetone, 130 °C, 60 min or 140 °C, 30 min, giving TBS-9f, h. (2) TBAF, THF, room temp., giving 9a–h in 14–82% yield. | ||
By elongation of the P1′ side chain the S3′ pocket in the enzyme can be reached.34 The initial attempts with the Suzuki–Miyaura reactions revealed that higher isolated yields of the P1′ extended inhibitors were obtained when having the tertiary alcohol TBS-protected during the cross-couplings. Therefore, derivative 8, synthesised according to the conditions mentioned in Scheme 1 except for the last deprotection step, was used as the aryl palladium precursor in these reactions. Controlled microwave heating of sealed reaction mixtures containing 1 equiv. aryl bromide 8, 3.0 equiv. of the boronic acid reagent, 3.0 equiv. of K2CO3 as the base, 0.05 equiv. of Herrmann's palladacycle35 as the Pd-source and 0.10 equiv. of the (t-Bu)3P releasing salt [(t-Bu)3PH]BF436 in DME/H2O at 120 °C for 20 min (Method A)31,37 rendered complete conversion of the starting material. TBAF-mediated deprotection and purification, either on silica gel or with RP-HPLC, gave products 9a–e in moderate to high isolated yields (Table 2).
| Cmpd | R′= | Yieldb (%) | K i/nM | EC50/μM | CC50/μM |
|---|---|---|---|---|---|
| a Conditions: see Schemes 2 (9a–h) and 3 (13a–b). b Isolated yield in the coupling/deprotection step. c Microwave conditions: Suzuki–Miyaura coupling (Method A). d Sonogashira coupling (Method C). e Sonogashira coupling (Method B). f P app (Caco-2) = 21 × 10−6 cm s−1, CLint = >300 μL min−1 mg−1. g P app (Caco-2) = <1 × 10−6 cm s−1, CLint = 63 μL min−1 mg−1. h P app (Caco-2) = 1.9 × 10−6 cm s−1, CLint = 20 μL min−1 mg−1. i P app (Caco-2) = 6.1 × 10−6 cm s−1, CLint = >300 μL min−1 mg−1. | |||||
| C | — | — | 2.8 | 0.17 | |
| 9a c , f |
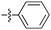
|
44 | 4.6 | 1.0 | >50 |
| 9b c , g |
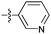
|
82 | 3.1 | 1.0 | >50 |
| 9c c , h |
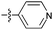
|
73 | 3.5 | 1.1 | >50 |
| 9d c |
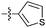
|
21 | 3.6 | 1.0 | >50 |
| 9e c |
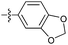
|
35 | 4.8 | 1.2 | >50 |
| 9f d |
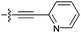
|
30 | 7.0 | 1.2 | >50 |
| 9g e , i |
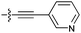
|
14 | 3.4 | 2.1 | >50 |
| 9h d |
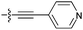
|
29 | 6.6 | 8.9 | >50 |
| 13a |
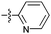
|
67 | 6.3 | 4.9 | >50 |
| 13b |
|
75 | 3.5 | 3.7 | >50 |
To be able to incorporate ethynyl-pyridines as P1′ substituents, Sonogashira-coupling chemistry was used with PdCl2(PPh3)2 as the catalyst.38 A microwave induced copper mediated Sonogashira reaction at 130 °C for 60 min (Method B)39 provided 9g in a disappointing yield of 14%.
In an effort to improve the outcome, different Sonogashira protocols were evaluated14,40–42 and a microwave heated copper-free cross-coupling methodology, inspired by the procedures reported by Shi and Zhang42 and Dieck and Heck,43 was chosen to produce 9f and 9h.
Using 1 equiv. aryl bromide 8, 2.5 equiv. ethynyl–pyridine, 0.1 equiv. PdCl2(PPh3)2, 4 equiv. piperidine in a water/acetone mixture (3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 140 °C for 30 min or 130 °C for 60 min (Method C), and subsequent deprotection, inhibitors 9f and 9h were synthesised and isolated in moderate yields (Table 2).
:1) at 140 °C for 30 min or 130 °C for 60 min (Method C), and subsequent deprotection, inhibitors 9f and 9h were synthesised and isolated in moderate yields (Table 2).
In the case of the 2-pyridyl and 2-thiazole substituents, the problem with competing protodeboronation and the unavailability of the boronic acid reactants at the time rendered us to produce these analogs according to an alternative approach (Scheme 3). Starting from the commercially available aldehydes 10a and 10b, and the hydrazide 11 (prepared according to a literature procedure21), β-nitrogen benzylated compounds 12a and 12b were synthesised by reductive amination at room temperature using NaCNBH3. Coupling between these hydrazides and the Dess–Martin oxidised aldehyde 5a, followed by deprotection of the tertiary alcohol, resulted in the desired inhibitors 13a and 13b in good isolated yields (Scheme 3).
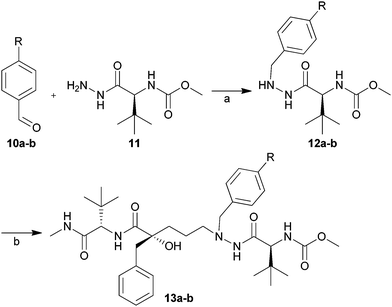 | ||
| Scheme 3 Synthesis of inhibitors 13a and 13b. Reagents and conditions: (a) TsOH, acetic acid, NaCNBH3, THF, room temp. Isolated yields: 12a, 64% and 12b, 60%; (b) (1) Dess–Martin periodinane, DCM, room temp; (2) 12a or 12b, Na(OAc)3BH, AcOH, THF, room temp; (3) TBAF, THF, room temp. Isolated yields: 13a 67% and 13b 75%. | ||
When optimising the P1′ side chain for antiviral activities (summarised as Ki and EC50 values in Table 2), no significant improvement in the biological activity was seen compared to 7a. The best inhibitor in the series (9b, Ki = 3.1 nM, EC50 = 1.0 μM) was almost equipotent to the indanol amide-containing reference compound C in the enzyme assay.
The heteroaromatic extensions of the P1′-group in inhibitors 9b–d resulted in slightly lower Ki values than those of inhibitors 9a and 9e (Table 2). Further elongations, as in 9f–h, were tolerated but not beneficial. The 3-pyridyl and 3-ethynyl pyridyl P1′-substituents (9b and 9g) provided improved inhibitory potencies compared with their 2-pyridyl analogues (13a, 9f) and equal or improved potencies compared with their 4-pyridyl analogues 9c and 9h, respectively.
In the cell assay, compounds 9a–f all produced EC50-values around 1 μM. The 4-ethynyl pyridyl analogue (9h) had only one ninth of the antiviral activity, compared to the best inhibitor of the series (9a). Inhibitors 13a and 13b lost five and four fold, respectively, compared to 9a.
Permeability (Caco-2) and stability (CLint) studies were performed for inhibitors 7a, 9a, 9b, 9c and 9j. When comparing permeability and stability data for 7a (Papp = 4.6 × 10−6 cm s−1, CLint = 180 μL min−1 mg−1) with Atz (Papp = 5.3 × 10−6 cm s−1, CLint = 90 μL min−1 mg−1),15 Wempe et al. recently reported CLint = 140 μL min−1 mg−1 for Atz,447a showed results in the same range as Atz. Varying the P1′ as in 9a gave high permeability but unfortunately also decreased stability (Papp = 21 × 10−6 cm s−1, CLint = >300 μL min−1 mg−1). With pyridyls in P1′ (9b and 9c) more stable compounds were achieved (CLint = 63 μL min−1 mg−1 and 20 μL min−1 mg−1 respectively) but at the cost of reduced permeability (Papp = <1 × 10−6 cm s−1 and Papp = 1.9 × 10−6 cm s−1, respectively).
X-Ray structure analysis
A number of active inhibitors were selected for co-crystallisation with a drug-resistant variant of HIV-1 protease containing the mutations Leu63Pro, Val82Thr and Ile84Val.45 The structures of the complexes between the protein and compounds 9a (PDB code 2xye) and 9d (PDB code 2xyf) were solved and refined to 2.0 Å and 1.8 Å resolution, respectively. Overviews of the binding patterns are shown in Fig. 2. Comparisons were made with the structures of 14 (with a (1S,2R)-1-amino-2-indanol in the P2 position, PDB code 2uxz)17 and Atz (PDB code 3EL9).As expected, the compounds 9a and 9d bind to the protease with the same overall conformation as 14 (Fig. 2). A complicating factor, for the comparisons of the inhibitor complexes, was the fact that compounds 9a and 9d were rotated 180° as compared to compound 14 and Atz.29,46,47
The most apparent difference in binding pattern to the protease active site between the four compounds is the positioning over the two catalytic aspartate residues.46 Atz places the central hydroxyl symmetrically over the two catalytic aspartate residues, while compounds 9a, 9d and 14 position the corresponding tertiary hydroxyl group in an asymmetric fashion, as a consequence of the larger central motif (Fig. 2). As a result of this asymmetric arrangement, the interactions with the catalytic aspartates (Asp25/125) are less developed. Thus, the hydrogen bonds are fewer and weaker.
The structural water, chelated by the central carbonyl groups and binding the flap Ile50 and Ile150 main-chain nitrogens, is conservatively positioned in all four structures with bond distances of 2.6–3.0 Å.
Atz, 9a and 9d all participate in a hydrogen bond to Gly48/148 (2.9–3.0 Å) which is not possible from the indanol amide of inhibitor 14.
Due to the longer central carbon tether of 9a and 9d the P1′ diaryl side chains come close enough to Phe53/153 to form edge-on aromatic interactions, and furthermore forces the residue and Phe53/153 to accommodate for the larger P1′ group (Fig. 2 and 3).
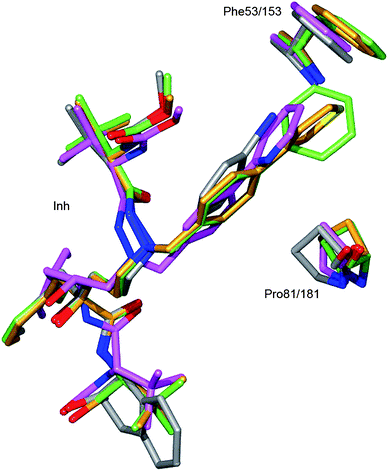 | ||
| Fig. 3 Analysis of the effect of the extended core structure of the positioning of P1′ side chain. Compound 9a and its interacting residues are in green, 9d in gold, 14 in gray and Atz in magenta. The distances between the P1′ side chain and Phe53/153 in 9a and 9d are 3.3 Å and 3.5 Å respectively. For Atz and 14 the corresponding distances are 4.0 Å and 4.4 Å respectively. In order to accommodate for the contact with the P1′ side chains of 9a and 9d the residue Phe53/153 side chain was moved approximately 1–1.5 Å in these complexes. The mobility of the flap structure is further demonstrated in that also the main chain atoms at Phe53/153 have moved approximately 0.5 Å in the 9a and 9d complexes compared to the Atz complex. Also Pro81/181 has moved (0.7–1.2 Å) due to the interaction with 9a and 9d compounds. | ||
Neither the 2-pyridyl containing P1′ of Atz nor the para-bromo phenyl in P1′ of 14 reach Phe53/153. A detailed analysis of effects of the different P1′ substitutions is shown in Fig. 3.
The Pro81/181 counts as part of both the S1/S1′ and the S3/S3′ pockets.48 The elongated P1′ side chains in inhibitor 9a and 9d facilitate an improved number of hydrophobic interactions with Pro81/181 within 3.2–3.6 Å. To make room for the elongated P1′ side chains of 9a and 9d, Pro81/181 has moved (0.7–1.2 Å) compared to 14 (Fig. 3).
The HIV-1 protease's ability to accommodate different natural target sequences45,49,50 is also reflected in its binding to the different inhibitor structures.51 Thus the flap residues at S2 adopt to accommodate for the bulky tert-leucine group and likewise for the flat but larger indanol group (Fig. 4).
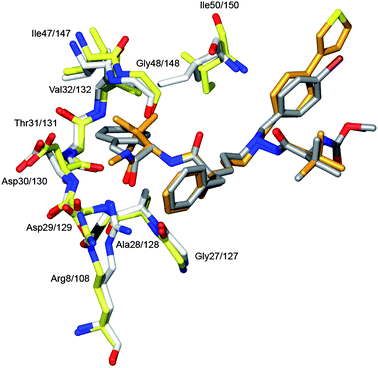 | ||
| Fig. 4 Flexible accommodation of the protein side- and main-chain atoms to the size variations of the inhibitor structures. This property of the protease molecule is exemplified here by the structures of the 9d and 14 complexes. In gold and yellow is shown 9d and the corresponding protein residues; likewise in grey and white 14. The bulky tert-leucine moiety in 9d forces the flap residues Ile147/147 and Gly48/148 to move 0.6 Å compared to their position when co-crystallised with 14. Even larger shift is observed at Arg8/108 which shifts its position with about 2.0 Å between the two complexes. In the complex with 14 the Arg8/108 Cζ and NH atoms close-packs at 3.6 Å distance with an edge-on cation–π interaction52 to the P1 of the phenyl group, whereas in the complex with 9d the Arg8/108 is pushed away by the P3 methyl amide group. | ||
Though Arg8/108 is involved in hydrogen bonding, it can move as much as 2 Å to optimise binding to 9d and 14, respectively.
Discussion
The biological evaluation of the P2–P3 diversified compounds is summarised in Table 1. Compound 7c (Ki = 2.7 nM) was the most potent compound in the enzyme assay, equipotent to reference compound C, and slightly better than indanol-compound 14.17Both 7a and 7c showed a twofold reduction in potency in the cell-based assay (EC50 = 2.0–2.1 μM) compared to analogously substituted indanol amide-compound (EC50 = 0.85 μM),17 and a tenfold reduction in potency compared to the P1′-optimised compound C. This series was less affected by the substitution of the indanol amide P2 moiety compared to series A where similar substitution gave more than 33-fold loss in potency.13
Inhibitors 7b and 7e, having the longest side chains in the P2 position, seem to be too large for the S2 pocket as reflected by a decreased potency compared to 7a, 7c and 7d. The attempt to conserve the H-bond donating capacity to Asp29/129 in 7f was not advantageous, probably due to the lost interactions with the backbone of Gly48/148 and the truncation which leaves the P2–P3 pocket unoccupied.
Variations of the P1′ group of 7a had little impact on the enzymatic inhibition but resulted in improved activity in the cell culture assay, compared to the para-bromo compound, 7a (Table 2). Only two to five times higher inhibitor concentrations were needed to reach the EC50 value of the metabolically unstable but highly potent indanol-amide compound 14.17
The Sonogashira promoted ethynyl–pyridyl elongations of the P1′ generating 9f–h were well tolerated by the enzyme but did not improve the enzyme inhibition or antiviral activity compared to the para-aryl substituted compounds. The 3- and 4-ethynylpyridyles 9g–h did not seem to be beneficial for inhibition.
Replacement of the previously used indanol amide P2 group14,15,17 generated some conformational changes within the protease (Fig. 4). However, the valine and tert-leucine P2 substituents seem to fill the available space in the S2 pocket and were well tolerated. Hence, they are suitable alternatives to the indanol moiety. If the size of the P2-group was increased, as in 7e, or reduced too much, as in 7d, a lower activity on both enzyme and in cell based assay was observed.
When truncating the P2–P3 region, as in 7f, all antiviral cell activity was lost, probably due to reduced permeability and loss of interactions with Gly49/149 and Asp29/129.
With their elongated backbone, the new series of inhibitors fitted well in the enzyme, even compared to Atz, as seen in Fig. 2. Inhibitors 9a and 9d showed edge-on interactions with Phe53/153 (Fig. 3) a finding which is observed neither in the case of 14, nor for Atz.
The Arg8/108 was forced to move by the methyl amide P3 group and the edge-on cation–π interactions between Arg8/108 and the Phe P1 group were disrupted. Nevertheless, the antiviral activity was not notably decreased by this change (9dKi = 3.6 nM, 14Ki = 3.3 nM17).
The 9 and 13 series presented herein show example of a successful replacement of the previously used indanol amide in P2, known for its good permeability, with the tert-butyl methyl amide and still furnishing in molecules with medium (9j) to high permeability (7a, 9a).
Conclusion
Herein, we have presented the synthesis, evaluation, and X-ray structural analysis of a series of tertiary alcohol containing HIV-1 proteases inhibitors, in which the previously used P2–P3-indanol amide fragment was replaced with aliphatic and aromatic amino acidN-methyl amide moieties. When using the tert-leucine and valine-derived P2–P3 fragments, good HIV-1 protease inhibition in agreement with the results using the metabolically unstable indanol amide were obtained. After optimisation of the P1′ side chain, low micromolar EC50 values were measured. The series also furnished examples with permeability and stability data that correspond well with data from Atz. Co-crystallisation of inhibitors 9a and 9d with the HIV-1 protease revealed that the tert-leucine methyl amide moiety fitted well in the S2–S3 pocket, partly due to an expansion of the S3 pocket. The tertiary alcohol generated non-symmetrical hydrogen bonds with the catalytic aspartates (Asp25 and Asp125) and novel edge-on interactions between the P1′ groups in 9a and 9d with Phe53/153 were detected.Experimental section
General procedure A: synthesis of lactone 4
Dry DCM (15–40 mL) was added to the mixture of acid 2 (1.0 equiv.), amino acid methyl amide 3 (1.0 equiv.), EDC (1.1 equiv.) and HOBt (1.1 equiv.). The mixture was stirred for 1 h at room temperature. The reaction was quenched with 30 cm3water, filtered and extracted with 2 × 30 cm3DCM (or ethyl acetate). The organic layers were combined, dried over MgSO4, concentrated and purified on silica gel to give 4 in 16–47% yield.General procedure B: synthesis of 5
To the solution of 4 (1.0 equiv.) in 15 cm3diethyl ether, LiBH4 (3.0 equiv.) was added at room temperature. The reaction mixture was stirred until full conversion and thereafter quenched with NH4Cl, followed by extraction with 3 × 30 cm3EtOAc. The combined organic layers were dried with MgSO4 and concentrated to give the crude diol. The resulting diol was dissolved in 4 cm3 dry pyridine, then 5.0 equiv. trimethylacetyl chloride was added to the pyridine solution and stirred at room temperature for 1.5 h (sometimes overnight). Thereafter, 15 cm3water was added to the mixture, which was extracted with 3 × 15 cm3ether, dried with MgSO4 and concentrated to give the crude monoester. Next, 15 cm3 dry DCM and triethylamine (6.0 equiv.) were added to the intermediate. After 5 min tert-butyl dimethyl silyltriflate (TBSOTf) (3.0 equiv.) was added at 0 °C, and the mixture was stirred at room temperature for 3 h or overnight. The solution was concentrated, extracted with diethyl ether and water. The organic layer was dried with MgSO4, filtered and concentrated to give a crude oily intermediate. The intermediate was subsequently dissolved in 15 cm3diethyl ether and LiBH4 (3.0 equiv.) was added to the solution at room temperature. Portions of LiBH4 were then added every 2 h until full conversion was achieved. The reaction was quenched with saturated aqueous NH4Cl solution and thereafter extracted with 3 × 15 cm3ether. The combined organic layers were dried over MgSO4, concentrated and purified on silica gel with 30–100% EtOAc in petroleum ether bp 40–60 °C to give the title compound 5 in 25–85% yield.General procedure C: synthesis of 7
To the solution of compound 5 (1.0 equiv.) in 5 cm3 dry DCM Dess–Martin reagent (1.1–1.2 equiv.) was added and the mixture was stirred at room temperature for about 40 min. Thereafter 4 cm3 saturated NaHCO3 solution and 4 cm3 saturated Na2S2O3 solution were added and the mixture was extracted with 3 × 15 cm3DCM. The combined organic layers were dried with MgSO4 and concentrated to give the aldehyde. Then {(S)-1-[N′-(4-bromo-benzyl)-hydrazinocarbonyl]-2,2-dimethyl-propyl}-carbamic acid methyl ester (6) (0.5 equiv.) and 10 cm3THF were added to the mixture. Acetic acid (1.0–1.5 equiv.) was added to the solution and the mixture was stirred for 15 min at room temperature. Thereafter Na(OAc)3BH (1.5–3.0 equiv.) was added and the mixture was stirred overnight. The reaction was quenched with saturated aqueous solution of NH4Cl and extracted with 3 × 15 cm3DCM. The combined organic layers were dried with MgSO4, concentrated and purified on silica gel with 50–100% EtOAc in petroleum ether (bp 40–60 °C). The fractions were combined and concentrated under reduced pressure. The protection group was removed using 10.0 equiv. 1.0 M TBAF in THF overnight at room temperature. The reaction mixture was purified with EtOAc or 5% MeOH in DCM on silica gel and freeze dried to give product 7 in 3–81% yield.Acknowledgements
We thank the Swedish Research Council (VR) and the Swedish Foundation for Strategic Research (SSF) for financial support, upper-secondary school student Mr Daniel Holmberg for production of the crystals and master thesis student Helena Löfgren for contributing with laborative work with 7f.Notes and references
- A. Brik and C.-H. Wong, Org. Biomol. Chem., 2003, 1, 5–14 RSC.
- J. T. Randolph and D. A. DeGoey, Curr. Top. Med. Chem., 2004, 4, 1079–1095 CrossRef CAS.
- C. Armbruster, Anti-Infect. Agents Med. Chem., 2008, 7, 201–214 Search PubMed.
- J. Pokorna, L. Machala, P. Rezacova and J. Konvalinka, Viruses, 2009, 1, 1209–1239 Search PubMed.
- R. K. Ghosh, S. M. Ghosh and S. Chawla, Expert Opin. Pharmacother., 2011, 12, 31–46 Search PubMed.
- F. Rodríguez-Barrios and F. Gago, Curr. Top. Med. Chem., 2004, 4, 991–1007 CrossRef CAS.
- F. Clavel and A. J. Hance, N. Engl. J. Med., 2004, 350, 1023–1035 CrossRef CAS.
- Y. Mehellou and E. De Clercq, J. Med. Chem., 2010, 53, 521–538 CrossRef CAS.
- A. M. J. Wensing, N. M. van Maarseveen and M. Nijhuis, Antivir. Res., 2010, 85, 59–74 Search PubMed.
- E. De Clercq, Int. J. Antimicrob. Agents, 2009, 33, 307–320 Search PubMed.
- T. Hawkins, Antivir. Res., 2010, 85, 201–209 Search PubMed.
- A. K. Ghosh, B. D. Chapsal, A. Baldridge, M. P. Steffey, D. E. Walters, Y. Koh, M. Amano and H. Mitsuya, J. Med. Chem., 2011, 54, 622–634 Search PubMed.
- J. K. Ekegren, J. Gising, H. Wallberg, M. Larhed, B. Samuelsson and A. Hallberg, Org. Biomol. Chem., 2006, 4, 3040–3043 RSC.
- J. K. Ekegren, N. Ginman, A. Johansson, H. Wallberg, M. Larhed, B. Samuelsson, T. Unge and A. Hallberg, J. Med. Chem., 2006, 49, 1828–1832 CrossRef CAS.
- J. K. Ekegren, T. Unge, M. Z. Safa, H. Wallberg, B. Samuelsson and A. Hallberg, J. Med. Chem., 2005, 48, 8098–8102 CrossRef CAS.
- A. K. Mahalingam, L. Axelsson, J. K. Ekegren, J. Wannberg, J. Kihlstrom, T. Unge, H. Wallberg, B. Samuelsson, M. Larhed and A. Hallberg, J. Med. Chem., 2010, 53, 607–615 Search PubMed.
- X. Wu, P. Öhrngren, J. K. Ekegren, J. Unge, T. Unge, H. Wallberg, B. Samuelsson, A. Hallberg and M. Larhed, J. Med. Chem., 2008, 51, 1053–1057 Search PubMed.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CAS.
- S. K. Balani, B. H. Arison, L. Mathai, L. R. Kauffman, R. R. Miller, R. A. Stearns, I. W. Chen and J. H. Lin, Drug Metab. Dispos., 1995, 23, 266–270 Search PubMed.
- J. H. Lin, Adv. Drug Delivery Rev., 1999, 39, 33–49 CrossRef CAS.
- G. Bold, A. Fässler, H.-G. Capraro, R. Cozens, T. Klimkait, J. Lazdins, J. Mestan, B. Poncioni, J. Rösel, D. Stover, M. Tintelnot-Blomley, F. Acemoglu, W. Beck, E. Boss, M. Eschbach, T. Hurlimann, E. Masso, S. Roussel, K. Ucci-Stoll, D. Wyss and M. Lang, J. Med. Chem., 1998, 41, 3387–3401 CrossRef CAS.
- A. Fässler, G. Bold, H.-G. Capraro, R. Cozens, J. Mestan, B. Poncioni, J. Rösel, M. Tintelnot-Blomley and M. Lang, J. Med. Chem., 1996, 39, 3203–3216 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-I. Negishi, Wiley-Interscience, New York, 2002, vol. 1, pp. 249–262 Search PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 874–922 Search PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- X-Ray of (S)-4a is included in ESI†.
- A. Wlodawer and J. W. Erickson, Annu. Rev. Biochem., 1993, 62, 543–585 CrossRef CAS.
- B. Mahalingam, Y. F. Wang, P. I. Boross, J. Tozser, J. M. Louis, R. W. Harrison and I. T. Weber, Eur. J. Biochem., 2004, 271, 1516–1524 Search PubMed.
- M. Larhed and A. Hallberg, J. Org. Chem., 1996, 61, 9582–9584 CrossRef CAS.
- J. Wannberg, K. Ersmark and M. Larhed, in Topics in Current Chemistry, SpringerBerlin, Heidelberg, 2006, pp. 167–198 Search PubMed.
- B. S. Robinson, K. A. Riccardi, Y.-F. Gong, Q. Guo, D. A. Stock, W. S. Blair, B. J. Terry, C. A. Deminie, F. Djang, R. J. Colonno and P.-F. Lin, Antimicrob. Agents Chemother., 2000, 44, 2093–2099 Search PubMed.
- H. O. Andersson, K. Fridborg, S. Löwgren, M. Alterman, A. Muhlman, M. Björsne, N. Garg, I. Kvarnström, W. Schaal, B. Classon, A. Karlen, U. H. Danielsson, G. Ahlsen, U. Nillroth, L. Vrang, B. Oberg, B. Samuelsson, A. Hallberg and T. Unge, Eur. J. Biochem., 2003, 270, 1746–1758 Search PubMed.
- W. A. Herrmann, C. Brossmer, K. Ofele, C. P. Reisinger, T. Priermeier, M. Beller and H. Fischer, Angew. Chem., Int. Ed. Engl., 1995, 34, 1844–1848 CrossRef CAS.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295–4298 CrossRef CAS.
- J. Wannberg, D. Dallinger, C. O. Kappe and M. Larhed, J. Comb. Chem., 2005, 7, 574–583 CrossRef CAS.
- Y. Liang, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2006, 71, 379–381 CrossRef.
- M. Erdelyi and A. Gogoll, J. Org. Chem., 2001, 66, 4165–4169 CrossRef CAS.
- J. Wannberg, Y. A. Sabnis, L. Vrang, B. Samuelsson, A. Karlen, A. Hallberg and M. Larhed, Bioorg. Med. Chem., 2006, 14, 5303–5315 Search PubMed.
- K. Ersmark, I. Feierberg, S. Bjelic, E. Hamelink, F. Hackett, M. J. Blackman, J. Hulten, B. Samuelsson, J. Qvist and A. Hallberg, J. Med. Chem., 2004, 47, 110–122 CrossRef CAS.
- S. Shi and Y. Zhang, Synlett, 2007, 1843–1850 CAS.
- H. A. Dieck and F. R. Heck, J. Organomet. Chem., 1975, 93, 259–263 CrossRef.
- M. F. Wempe and P. L. Anderson, Drug Metab. Dispos., 2011, 39, 522–527 Search PubMed.
- I. T. Weber and J. Agniswamy, Viruses-Basel, 2009, 1, 1110–1136 Search PubMed.
- M. A. Navia, P. M. D. Fitzgerald, B. M. Mckeever, C. T. Leu, J. C. Heimbach, W. K. Herber, I. S. Sigal, P. L. Darke and J. P. Springer, Nature, 1989, 337, 615–620 CrossRef CAS.
- P. Bagossi, Y. S. E. Cheng, S. Oroszlan and J. Tozser, Protein Eng., 1998, 11, 439–445 Search PubMed.
- K. Appelt, Perspect. Drug Discovery Des., 1993, 1, 23–48 Search PubMed.
- M. Prabu-Jeyabalan, E. Nalivaika and C. A. Schiffer, Structure, 2002, 10, 369–381 CrossRef CAS.
- J. Anderson, C. Schiffer, S.-K. Lee and R. Swanstrom, Handb. Exp. Pharmacol., 2009, 189, 85–110 Search PubMed.
- A. Ali, R. M. Bandaranayake, Y. F. Cai, N. M. King, M. Kolli, S. Mittal, J. F. Murzycki, M. N. L. Nalam, E. A. Nalivaika, A. Ozen, M. M. Prabu-Jeyabalan, K. Thayer and C. A. Schiffer, Viruses, Basel, 2010, 2, 2509–2535 Search PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00077b |
| This journal is © The Royal Society of Chemistry 2011 |