Nitro-based inhibitors against thermolysin†
Guan Rong
Tian
a,
Si-Hong
Wang
a,
Shou-Feng
Wang
a,
Ling-Qiang
Meng
a,
Hui
Li
a,
Zong-Hao
Zeng
b and
Jing-Yi
Jin
*a
aKey Laboratory of Natural Resources of Changbai Mountains & Functional Molecules of Ministry of Education, Gongyuan Raod 977, Yanji city, Jilin Prov 133002, China. E-mail: jyjin-chem@ybu.edu.cn; Fax: +86-433-2732456; Tel: +86-433-2732286
bInstitute of Biophysics, Chinese Academy of Science, Beijing, 100871, China
First published on 17th May 2011
Abstract
A series of nitro-based compounds were synthesized and evaluated as inhibitors against thermolysin (TLN). The binding modes of the nitro group at the active site of TLN were characterized based on the structural analysis of TLN-(R)-2-benzyl-3-nitropropionic amide complex, which was suggested as a pseudo-transition state analogue. Further structure–activity relationship studies disclosed that both the stereochemistry of the P1 chains and the bulkiness of the P2′ substituents were crucial to the potent inhibition of TLN.
Thermolysin (TLN, E.C. 3.4.24.27) is a heat-stable proteolytic enzyme isolated from Bacillus thermoproteolyticus and carries a catalytically essential zinc ion at the active site. It is an extracellular endopeptidase, hydrolyzing the peptide bond on the imino side of the amino acid residue having a large hydrophobic side chain, such as Leu, Ile, and Phe.1TLN is one of the prototypical zinc proteases, and has played an important role as a model enzyme for the development of inhibitors for other zinc proteases of medicinal interest such as angiotensin I-converting enzyme (ACE, E.C. 3.4.15.1) and neprilysin (NEP, E.C. 3.4.24.11).2
The most popular approach to designing inhibitors of zinc proteases is the introduction of a zinc binding group (ZBG) in an optimized peptidic or non-peptidic scaffold.3 However, it is still a challenge to obtain potent inhibition based on the known ZBG for a specific enzyme. For example, in the inhibition of a neutral proteinase isolated from Saccharomonospora canescens (NPS),4 which shares many common features with TLN, a phosphoramidon-based inhibitor is very weak with an inhibitory constant (Ki) value of 65.0 μM but potent inhibition was observed for TLN (Ki = 0.034 μM).5 It is further complicated by the stereochemistry of inhibition. 2-Benzylsuccinic acids are inhibitors for TLN as well as carboxypeptidase A (CPA, E.C. 3.4.17.1), the other zinc-dependent enzyme whose active site structure, mechanism of action, and stereospecificity resemble those of TLN.1a,6 The L-form of BSA binds CPA much more tightly than its enantiomer,7 while the D-form is the preferred inhibitor for TLN accompanied with the different coordinative modes to the zinc ion at the active site.6c
Being an isostere of the carboxylate, the nitro group has been investigated in various enzyme–inhibitor complexes.8 A nitro-based inhibitor for CPA, L-2-benzyl-3-nitrosuccinic acid, exhibits slightly improved inhibitory activities (Ki = 0.15 μM) compared with L-BSA (Ki = 0.45 μM).8b However, nitro-based inhibitors have not been reported previously. As mentioned above, a practical investigation is necessary to develop nitro-based TLN inhibitors. Our work was thus devoted to exploring the binding modes of the nitro group at the active site of TLN as well as to obtaining potent inhibition of TLN by the optimization of the side chains.
To examine the stereochemistry in the inhibition of TLN, 2-benzyl-3-nitropropionic amides (1) should be firstly screened as inhibitors for TLN due to their simple structure. In our opinion, their complexes with TLN could also provide enough structural information to disclose the binding modes of nitro group to the zinc ion at the active site of TLN. All optically active forms of 1 were then synthesized. For the dipeptide inhibitors against TLN, it is generally found that either benzyl or isobutyl was the optimized side chain for P1′ or P2′.9 We thus further synthesized four dipeptides 2–5 (Scheme 1). An alternative route to obtain optically active 3 and 5 involved first formation of the peptide bond, followed by bromination through the mesylate.10 The two diastereomers obtained could be separated by silica gel column chromatography with a mixture of n-hexane and ethyl acetate as eluent. Final introduction of the nitro group gave the optically active forms of 3 or 5 (Scheme 2). The stereochemical assignments for them were made on the basis of comparison of the [α] values with the compounds obtained using the first method. The corresponding configurations of the intermediates involved were thus correlated.
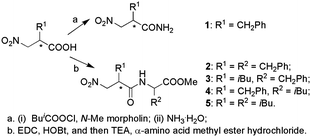 | ||
| Scheme 1 | ||
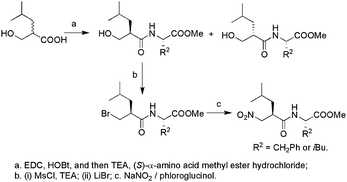 | ||
| Scheme 2 | ||
All compounds were assayed as competitive inhibitors for TLN using N-[3-(2-furyl)acryloyl]Gly-L-Leu-NH2 as substrate.9a Their Ki values were obtained from the respective plot for v0/v against the concentration of inhibitors and are collected in Table 1. Fig. 1 shows a Dixon plot for the inhibition of TLN.
| Compounds | K i (μM) |
|---|---|
| (±)-1 | 36 |
| (R)-1 | 7.6 |
| (S)-1 | 663 |
| (2′R, 2S)-2 | 0.95 |
| (2′S, 2S)-2 | 79 |
| (2′R, 2R)-2 | 1.40 |
| (2′S, 2R)-2 | 86 |
| (2′R, 2S)-3 | 0.84 |
| (2′S, 2S)-3 | 7.2 |
| (2′R, 2S)-4 | 0.37 |
| (2′S, 2S)-4 | 2.4 |
| (2′R, 2S)-5 | 0.083 |
| (2′S, 2S)-5 | 0.77 |
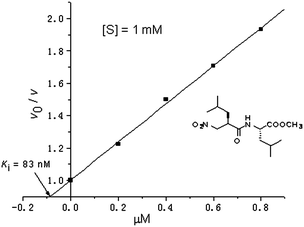 | ||
| Fig. 1 | ||
Firstly, consistent to L-specificity of TLN, (R)-1 showed more binding affinity to TLN than the (S)-form, which is different from the carboxylate-based inhibitor. TLN-(R)-1 crystals for X-ray diffraction were then obtained by soaking the enzyme crystals into the solution containing the racemic 1 and determined at a resolution of 1.5 Å (PDB ID code: 3FOR). The final model including all residues of TLN and (R)-1 was refined in the 38.48–1.93 Å (ESI†). Fig. 2 depicts the stereoview of the difference electron density in the region of the active site of TLN that is occupied by (R)-1. Distances of important interactions between TLN and (R)-1 in the complex are listed in Table 2.
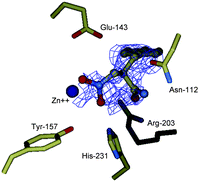 | ||
| Fig. 2 Difference electron density map for TLN-(R)-1 complex generated with Fourier coefficient |Fo| − |Fc| phases calculated from the final model omitting the bound inhibitor. | ||
| Enzyme residue | Contacting atom (inhibitors) and distance (Å) | |||||
|---|---|---|---|---|---|---|
| ZGPLL (5TMN) | Silanediol (1Y3G) | (R)-2 (3FOR) | ||||
| a The contact distances between the enzyme residues and the inhibitor atoms were measured according to the coordination files obtained from the Protein Data Bank. The PDB IDs of 5TMN,11a1Y3G11c and 3FOR correspond to the complexes of TLN with the inhibitor ZGPLL, silanediol and (R)-1, respectively. | ||||||
| Zn2+ | OP1 | 2.08 | OSi1 | 1.93 | ON1 | 2.25 |
| Zn2+ | OP2 | 3.01 | OSi2 | 3.28 | ON2 | 2.63 |
| His-231 NE2 | OP1 | 2.9 | OSi1 | 2.55 | ON1 | 3.11 |
| Glu-143 OE1 | OP2 | 2.5 | OSi2 | 2.76 | ON2 | 3.68 |
| Tyr-157 phenolic O | OP1 | 3.4 | OSi1 | 3.44 | ON1 | 3.96 |
| Asn-112 amide O | N amide | 3.09 | ||||
The electron density map for the TLN-(R)-1 complex shows that one molecule of (R)-1 is bound to the active site of TLN. Both oxygen atoms of the nitro group are involved in strong coordinative bonds to the zinc ion having the distances as 2.25 and 2.63 Å, respectively, which means that the nitro group also coordinates to the zinc ion in a bidentate mode (Table 2). One additional hydrogen bond between ON1 of (R)-1 and the NE2 of His-231 could be inferred, which is believed to be the oxyanion hole to stabilize the transition-state intermediate occurring in the TLN-catalyzed hydrolysis of the substrate.11 It is further found that the distance between ON2 of (R)-1 and the OE1 of Glu-143 is measured as 3.68 Å. The hydrogen bond of the nitro group with Glu-143 could not be thus expected. Compared to the generally accepted transition-state-analogue inhibitors of TLN,11a which are structurally featured by the gem-diolate oxygens stabilized by chelation to the zinc ion and two respective hydrogen bonds with Glu-143 and His-203, we thus proposed that the nitro group acts as a pseudo-transition-state-analogue. In addition, the amide nitrogen atom of (R)-1 formed a hydrogen bond with the amide oxygen atom of the side chain of Asn-112, which supported our design of the presented dipeptide compounds as TLN inhibitors.
Of the four optically forms of 2, (2′R, 2S)-2 is the most potent inhibitors for TLN. Considering the benzyl as the same side chain for both S1′ and S2′ hydrophobic pockets of TLN, it could be reasonably believed that the enzyme showed preference to the nitro-based dipeptides compounds with L,L-configurations. The stereochemistry of P1′ substituents seemed to have a great effect on the strong binding affinities to the enzyme, i.e., significant differences could be observed in comparison with (2′R, 2S)- and (2′S, 2S)-2, (2′R, 2R)- and (2′S, 2R)-2, respectively. Similarly, it was found that (2′R, 2S)-3, (2′R, 2S)-4, and (2′R, 2S)-5 are more than 7–9 fold more potent than (2′S, 2S)-3, (2′S, 2S)-4, and (2′S, 2S)-5, respectively. On the other hand, the right stereochemistry of the P2′ chains slightly enhanced the inhibitory activities of (2′R, 2S)-2 in comparison with (2′R, 2R)-2.
Replacement of the benzyl at the P1′ position of (2′R, 2S)-2 by an isobutyl group gave (2′R, 2S)-3 accompanied with a small improvement of the binding affinities to TLN. A similar tendency could be found in the P2′ position, where (2′R, 2S)-4 is about 3 fold more potent than (2′R, 2S)-2. It seemed that both the S1′ and S2′ pockets of TLN were preferred to the isobutyl group for the nitro-based dipeptide inhibitors. The most potent inhibition of TLN was achieved by (2′R, 2S)-5 with a Ki value of 83 nM, which was comparable to the silanediol-based inhibitor (Ki = 41 nM).9c The isobutyl as P2′ chain led to (2′R,2S)-5 more than 10-fold more potent than (2′R,2S)-3. It is thus reasonable to assume that the bulkiness of the P2′ chain more significantly affected the binding affinities of the dipeptides to TLN, that is, the isobutyl chain is better accommodated by the S2′ pocket of TLN than the benzyl group.
In summary, the nitro group as ZBG was introduced in the inhibition of TLN. Its binding modes at the active site of TLN were investigated on the basis of structural analysis of the enzyme–inhibitor complex, which was featured by a pseudo-transition state analog. Subsequent studies on the relationships between the inhibitory activities and the structures of the side chains disclosed that both the stereochemistry of the P1′ chain and the bulkiness of the P2′ chains mainly contributed to the potent inhibition of TLN. Among the present nitro-based compounds, (2′R, 2S)-N-(2-isobutyl-3-nitropropionyl)leucine methyl ester is the most effective inhibitor of TLN with a Ki comparable to the reported transition state analog inhibitor using silanediol as zinc-binding group.
Acknowledgements
The work was financially supported by the Science and Technology Committee of Jilin Province (No. 20060568), China.Notes and references
-
(a) B. W. Matthews, Acc. Chem. Res., 1988, 21, 333 CrossRef CAS
; (b) K. Inouye, S. B. Lee and B. Tonomura, Biochem. J., 1996, 315, 133 Search PubMed
-
(a) M. J. Wyvratt and A. A. Patchett, Med. Res. Rev., 1985, 5, 483
-
(a) F. E. Jacobsen, J. A. Lewis and S. M. Cohen, ChemMedChem, 2007, 2, 152 CrossRef CAS
- P. Dolashka, D. N. Georgieva, S. Stoeva, N. Genov, R. Rachev, A. Gusterova and W. Voelter, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1382, 207 Search PubMed
-
(a) L. H. Weaver, W. R. Kester and B. W. Matthews, J. Mol. Biol., 1977, 114, 119 CAS
-
(a) W. L. Lipscomb, Acc. Chem. Res., 1970, 3, 81 Search PubMed
- S. Mangani, P. Carloni and P. Orioli, J. Mol. Biol., 1992, 223, 573 CAS
-
(a) F. Sun, X. Huo, Y. Zhai, A. Wang, J. Xu, D. Su, M. Bartlam and Z. Rao, Cell, 2005, 121, 1043 Search PubMed
-
(a) J. Feder, L. R. Brougham and B. S. Wildi, Biochemistry, 1974, 13, 1186 Search PubMed
- H. S. Lee, J. D. Park and D. H. Kim, Bull. Korean Chem. Soc., 2003, 24, 467 Search PubMed
-
(a) H. M. Holden, D. E. Tronrud, A. F. Monzingo, L. H. Weaver and B. W. Matthews, Biochemistry, 1987, 26, 8542 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and full characterization of the presented compounds and kinetic assay and X-ray crystallography involved here. See DOI: 10.1039/c1md00088h |
| This journal is © The Royal Society of Chemistry 2011 |