Determination of structural requirements of influenza neuraminidase type A inhibitors and binding interaction analysis with the active site of A/H1N1 by 3D-QSAR CoMFA and CoMSIA modeling
Prashant R.
Murumkar
a,
Ly
Le
b,
Thanh N.
Truong
*b and
Mange Ram
Yadav
*a
aPharmacy Department, Faculty of Technology and Engineering, The M. S. University of Baroda, Kalabhavan. Vadodara, 390 001, Gujarat, India. E-mail: yadav-phar@msu.ac.in; Fax: +91 0265 2418927; Tel: +91 02645 2434187
bDepartment of Chemistry, The University of Utah, Salt Lake City, UT 84112, USA. E-mail: Troung@Chemistry.Utah.edu; Fax: +1 801 581-8433; Tel: +1 801 581-4301
First published on 31st May 2011
Abstract
As a basis for predicting structural features that may lead to the design of more potent and selective inhibitors of influenza neuraminidase type A, three-dimensional quantitative structure–activity relation (3D-QSAR) studies were performed on a set of sixty one known neuraminidase type A inhibitors. The studies include Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). Atom and centroid/atom based alignment and docked conformation-based alignments were used to develop different CoMFA models. A homology modelled A/H1N1 with one of the most active neuraminidase inhibitors (Relenza) inside the active site structure was used for docking purposes. The resulted docking based conformers of the inhibitors were used for developing the CoMFA model. The model developed by considering the docked conformation-based alignment was found to perform better than that developed by atom and centroid/atom based alignments with excellent predictive r2. The CoMFA model generated using docked conformer-based alignment served as alignment strategy for CoMSIA. The CoMSIA model with a combination of steric, electrostatic and acceptor fields yielded the highest cross-validated r2. CoMFA steric, electrostatic and CoMSIA donor contour maps were mapped in the active site of A/H1N1. These 3D-QSAR studies revealed indispensable structural features of different chemical classes of molecules which could be exploited for the structural modifications of these lead molecules in order to achieve improved neuraminidase type A inhibitory activity.
Introduction
Hemagglutinin and neuraminidase (NA), the two glycoproteins on the surface of the influenza virus, have long been considered as potential targets for the development of antiviral agents.1NA is responsible for viral release from infected cells and viral transport through the mucus in the respiratory tract.2NA, also called sialidase, cleaves terminal sialic acid residues from glycoconjugates, promoting the release of newly formed virus particles from infected cells. The three pathogenic viruses, the 1918 Spanish flu (H1N1), the 2003 Avian flu (H5N1) and the recent 2009 Swine flu (A/H1N1), share the same subtype 1 (N1) of the neuraminidase. Thus, influenza neuraminidase has been considered as an attractive target for the development of new drugs for influenza.1The 2009 outbreak of a new strain of influenza A virus subtype H1N1 generated concerns about a new pandemic. In the latter half of April 2009, the World Health Organization's pandemic alert level was sequentially increased from three to five and on 11 June 2009 the pandemic level had been raised to its highest level, level six. Dr Margaret Chan, Director-General of the World Health Organization (WHO), confirmed on 11 June 2009 that the H1N1 strain was indeed a pandemic, having nearly 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 confirmed cases worldwide.3 The current seasonal flu vaccine, which targets a different H1N1 strain, provides little or no protection against H1N1 pandemic. In terms of medication, the FDA approved two antiviral drugs, Tamiflu (oseltamivir) and Relenza (zanamivir), are effective against this new type of virus.4 Unfortunately, such virus types are known for their quick mutations and gene assortments which enable them to escape the host immune system and resist drugs. To be very specific, a case of swine flu resistant to Tamiflu was observed in Denmark on June 29, 2009 for the first time, and soon after that in Japan and Hong Kong.5 Thus, developing new antiviral drugs for this new H1N1 virus is a matter of extreme urgency. In this direction, this group has been actively engaged in research for developing new potential compounds to be used as A/H1N1 inhibiors.6,7
000 confirmed cases worldwide.3 The current seasonal flu vaccine, which targets a different H1N1 strain, provides little or no protection against H1N1 pandemic. In terms of medication, the FDA approved two antiviral drugs, Tamiflu (oseltamivir) and Relenza (zanamivir), are effective against this new type of virus.4 Unfortunately, such virus types are known for their quick mutations and gene assortments which enable them to escape the host immune system and resist drugs. To be very specific, a case of swine flu resistant to Tamiflu was observed in Denmark on June 29, 2009 for the first time, and soon after that in Japan and Hong Kong.5 Thus, developing new antiviral drugs for this new H1N1 virus is a matter of extreme urgency. In this direction, this group has been actively engaged in research for developing new potential compounds to be used as A/H1N1 inhibiors.6,7
Three-dimensional quantitative structure–activity relationship (3D-QSAR) has been considered as a powerful approach to gain insight into the relationship between the chemical structure and the biological activity. Our group has been involved in the computer aided designing of novel molecules of medicinal interest using 3D-QSAR.8–12 Considering the importance of developing newer antiviral drugs acting as A/Neuraminidase inhibitors and the challenges involved in discovering new drugs against fast mutating viruses caught our attention to first develop a reliable 3D-QSAR model for neuraminidase inhibitors containing different classes of compounds.
In the present study, 3D-QSAR analysis has been carried out on a series of multisubstituted pyrrolidine, cyclopentane- and cyclohexenecarboxylic acid derivatives as influenza neuraminidase type A inhibitors, in order to identify key structural elements required to design new drug candidates. The studies include both the 3D-QSAR techniques, Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). Docking based conformations of the selected compounds were used as one of the strategies to develop a highly predictive CoMFA model. The molecular model of H1N1 neuraminidase which was built by this group using H5N1 neuraminidase (PDB entry 2HU4) as the starting point, then mutating corresponding residues, was used for docking purpose. The detailed discussion on the H1N1 neuraminidase model can be found in our previous publication.6 These docked conformers were used to develop a 3D-QSAR model to get a better insight to design more potent neuraminidase inhibitors.
Results and discussion
CoMFA analysis
The negative logarithm of the IC50 (pIC50) was used as the dependent variable in the 3D-QSAR study (Tables 1–3). The PLS analysis results for all the three types of 3D-QSAR CoMFA modeling are summarized in Table 4. The CoMFA models were built using a training set of 47 compounds and tested using a test set of 14 compounds. Selection of the training set and test set molecules was done by considering the fact that test set molecules represented a range of biological activity similar to that of the training set. To diversify the training set and test set, they were grouped manually so that molecules possessing diverse functionalities and biological activity of wide range were included in both sets. The lower energy conformers obtained from the MULTISEARCH option in SYBYL were used in the study. All the molecules were aligned employing three different types of alignments. Various 3D-QSAR models were generated and based on the statistically significant parameters obtained, the best model was chosen.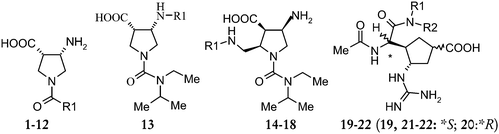
| Comp. | R1 | R2 | Biological activity | ||
|---|---|---|---|---|---|
| Observed | Predicted | ||||
| IC50/µM | pIC50 | pIC50 (CoMSIA) | |||
| 1 | –CH(C2H5)2 | — | 22 | 4.657 | 3.836 |
| 2 (T) | –N(C2H5)2 | — | 25 | 4.602 | 5.318 |
| 3 | –N(C3H7)2 | — | 32 | 4.494 | 4.064 |
| 4 |

|
— | 1.6 | 5.975 | 5.221 |
| 5 |
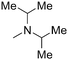
|
— | 4 | 5.397 | 5.396 |
| 6 (T) |
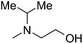
|
— | 21 | 4.677 | 5.079 |
| 7 |
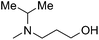
|
— | 2.1 | 5.677 | 5.558 |
| 8 |
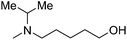
|
— | 2.0 | 5.698 | 5.541 |
| 9 (T) |

|
— | 19 | 4.721 | 5.790 |
| 10 |

|
— | 1.3 | 5.886 | 5.892 |
| 11 |

|
— | 46 | 4.337 | 5.46 |
| 12 |
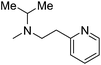
|
— | 1.3 | 5.886 | 5.891 |
| 13 (T) |
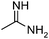
|
— | 13 | 4.886 | 5.229 |
| 14(T) | –COCH3 | — | 7.5 | 5.124 | 5.099 |
| 15 | –COC2H5 | — | 16 | 4.795 | 5.016 |
| 16 | –COCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
— | 96 | 4.017 | 5.005 |
| 17 | –COCF3 | — | 0.28 | 6.552 | 5.587 |
| 18 (T) | –SO2CH3 | — | 130 | 3.886 | 5.750 |
| 19 | H | CH(C2H5) | 0.06 | 7.221 | 7.595 |
| 20 | H | CH(C2H5) | 25.0 | 4.602 | 4.478 |
| 21 | –C2H5 | C2H5 | 0.07 | 8.154 | 7.670 |
| 22 | –C3H7 | C3H7 | 0.043 | 7.366 | 7.516 |

| Comp. | R′ | Biological activity | ||
|---|---|---|---|---|
| Observed | Predicted | |||
| IC50/nM | pIC50 | pIC50 (CoMSIA) | ||
| 23 | H | 6300 | 5.200 | 5.408 |
| 24 | CH3– | 3700 | 5.431 | 5.755 |
| 25 (T) | CH3CH2– | 2000 | 5.698 | 6.049 |
| 26 | CH3(CH2)2– | 180 | 6.744 | 6.349 |
| 27 | CH3(CH2)3– | 300 | 6.522 | 6.537 |
| 28 (T) | CH3(CH2)4– | 200 | 6.698 | 6.598 |
| 29 | CH3(CH2)5– | 150 | 6.823 | 6.618 |
| 30 | CH3(CH2)6– | 270 | 6.568 | 6.602 |
| 31 (T) | CH3(CH2)7– | 180 | 6.744 | 6.576 |
| 32 | CH3(CH2)8– | 210 | 6.677 | 6.550 |
| 33 | CH3(CH2)9– | 600 | 6.221 | 6.525 |
| 34 | (CH3)2CHCH2– | 200 | 6.698 | 7.682 |
| 35 (T) | CH3CH2(CH3)CH– | 10 | 8.00 | 7.280 |
| 36 | (s) CH3CH2(CH3)CH– | 9 | 8.045 | 8.055 |
| 37 |
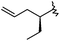
|
1 | 9.000 | 8.821 |
| 38 |
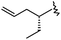
|
3 | 8.522 | 7.866 |
| 39 (T) |
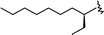
|
1 | 9.000 | 7.372 |
| 40 |
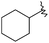
|
60 | 7.221 | 7.523 |
| 41 |
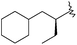
|
16 | 7.795 | 8.387 |
| 42 |
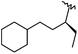
|
1 | 9.000 | 8.590 |
| 43 (T) |
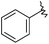
|
530 | 6.275 | 7.152 |
| 44 |
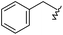
|
620 | 6.207 | 5.941 |
| 45 |
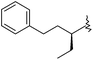
|
0.3 | 9.522 | 9.535 |
| 46 |
|
12 | 7.920 | 8.485 |
| 47 |

|
90 | 7.045 | 6.397 |
| 48 | H | 100 | 7.000 | 7.585 |
| 49 | CH3(CH2)2– | 2 | 8.698 | 8.681 |
| 50 | CH3(CH2)3– | 3 | 8.522 | 8.855 |
| 51 | (R)-CH3CH2(CH3)CH– | 0.5 | 9.301 | 8.971 |
| 52 (T) | (S)-CH3CH2(CH3)CH– | 0.5 | 9.301 | 8.922 |
| 53 | (CH3CH2)2CH– | 0.5 | 9.301 | 9.224 |
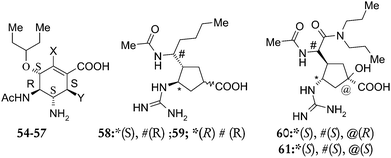
| Comp. | X | Y | Biological activity | ||
|---|---|---|---|---|---|
| Observed | Predicted | ||||
| IC50/µM | pIC50 | pIC50 (CoMFA) | |||
| 54 | H | H | 1 (nM) | 9.000 | 8.551 |
| 55 | CH3 | H | 2300 (nM) | 5.638 | 5.643 |
| 56 | F | H | 3 (nM) | 8.522 | 7.417 |
| 57 | H | CH3 | 1500 (nM) | 5.823 | 6.552 |
| 58 | — | — | 0.013 | 7.886 | 7.730 |
| 59 | — | — | 14.6 | 4.835 | 5.078 |
| 60 (T) | 4.9 | 5.309 | 5.130 | ||
| 61 | 2300 | 2.638 | 3.284 | ||
| Parameters | Alignment-Ic | Alignment-II | Alignment-III |
|---|---|---|---|
| R cv 2 | 0.415 | 0.483 | 0.519 |
| NC | 6 | 8 | 6 |
| R ncv 2 | 0.909 | 0.921 | 0.914 |
| SEE | 0.532 | 0.363 | 0.521 |
| F-value | 48.231 | 128.312 | 59.265 |
| R Pred 2 | 0.354 | 0.358 | 0.605 |
| Contribution (S; E) | 56.3; 44.7 | 59.6; 40.4 | 53.1; 46.9 |
A preliminary study was performed to study the importance of each field individually. All further analyses were performed with steric and electrostatic fields calculated at each grid point simultaneously. Partial least square (PLS) analysis was performed using varying column filtering values. Finally, column filtering was set to 2.0 kcal mol−1. Different alignments such as Alignments I to III were carried out as shown in Fig. 1.
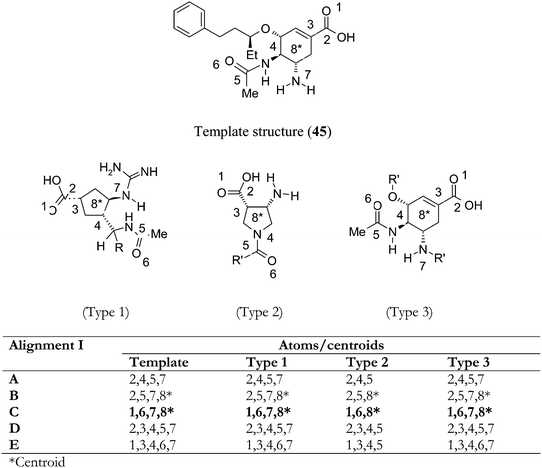 | ||
| Fig. 1 Designation of atoms for atom and atom–centroid based alignments for Method 1 (Alignments I A–E). | ||
Alignment I was carried out in five different ways (A–E) and various 3D-QSAR models were generated for all of them. Alignment Ic afforded the best statistical model hence only the Alignment Ic based model is discussed below. Alignment-Ic showed cross-validated r2 = 0.415 with six components, non-cross-validated r2 = 0.909, F-value 48.231, predictive r2 = 0.354; the steric and electrostatic contributions were 56.3 and 44.7%, respectively.
The CoMFA model generated from Alignment-II i.e. docking-based alignment showed cross-validated r2 = 0.483 with eight components, non-cross-validated r2 = 0.921, F-value 128.312, predictive r2 = 0.358 with 59.6 steric and 40.4% electrostatic contributions. Alignment-III (Table 4) yielded the highest cross-validated r2 of 0.519 with six components, non-cross-validated r2 of 0.914, F-value 59.265 and predictive r2 of 0.605 with 53.1% steric and 46.9% electrostatic contributions. The statistics in Table 4 shows that out of the three different CoMFA models developed on the basis of different alignments, the model developed with Alignment-III has the highest predictive power lending credit to the reliability of the active conformations obtained by Glide. The predicted pIC50 values are in good agreement with the experimental data with small residuals.
Quantitatively, the steric and electrostatic fields derived from the conventional atom–centroid fit molecular alignment (Alignment-Ic) is only ligand-centeric; whereas the one derived from the docking-based active conformation alignment (Alignment-III) is a combined product of the minimized conformations of the ligands and the active site structure. This innovative approach used in Alignment-III is predominantly responsible for the differences in the results of the two sets of CoMFA modeling. In the following discussion on CoMFA and CoMSIA analyses, Alignment-III only has been considered.
CoMSIA analysis
The CoMSIA analysis was performed using steric, electrostatic, hydrophobic, and hydrogen bond donor and acceptor fields as CoMSIA offers five different fields. 3D-QSAR models can be generated using the above fields in different combinations. Alignment-III used in CoMFA studies was used as the alignment for CoMSIA studies and the results of these studies are summarized in Table 5. The CoMSIA model showed considerable correlative and predictive properties. The CoMSIA model with the combination of all the fields yielded a cross-validated r2 = 0.446 with five components, non-cross-validated r2 = 0.913, F-value 64.035 and predictive r2 = 0.623. The contributions of steric, electrostatic, hydrophobic, hydrogen bond donor and acceptor fields of this model were 13.6, 19.7, 20.6, 21.5 and 24.5% respectively. The combination of steric, electrostatic and hydrogen bond donor fields yielded a CoMSIA model with a cross-validated r2 = 0.454 with four components, non-cross-validated r2 = 0.834, F-value 51.601 and predicted r2 = 0.619. The steric, electrostatic and hydrogen bond donor field contributions were 25.1, 35.5 and 39.3%, respectively.| Parameters | SE | SEH | SED | SEA | SEDA | SEHD | SEHDA |
|---|---|---|---|---|---|---|---|
| R cv 2 | 0.487 | 0.433 | 0.454 | 0.526 | 0.423 | 0.474 | 0.446 |
| N | 4 | 5 | 4 | 6 | 3 | 3 | 5 |
| R ncv 2 | 0.838 | 0.890 | 0.834 | 0.915 | 0.788 | 0.828 | 0.913 |
| SEE | 0.699 | 0.582 | 0.704 | 0.517 | 0.789 | 0.711 | 0.517 |
| F-Value | 52.836 | 64.795 | 51.601 | 70.215 | 51.990 | 69.674 | 64.035 |
| R Pred 2 | 0.617 | 0.589 | 0.619 | 0.641 | 0.628 | 0.618 | 0.623 |
| Contributions | 39.2; 60.8 | 22.4; 38.4; 39.2 | 25.1; 35.5; 39.3 | 25.1; 35.8; 39.1 | 17.5; 25.3; 27.6; 29.6 | 16.6; 26.3; 29.6; 27.5 | 13.6; 19.7; 20.6; 21.5; 24.5 |
The combination of steric, electrostatic and hydrophobic fields yielded a CoMSIA model with a cross-validated r2 = 0.433 with five components, non-cross-validated r2 = 0.890, F-value 64.795 and predicted r2 = 0.589. The steric, electrostatic and hydrophobic field contributions were 22.4, 38.4 and 39.2%, respectively. The combination of steric, electrostatic hydrogen bond donor and hydrogen bond acceptor fields yielded a CoMSIA model with a cross-validated r2 = 0.423 with three components, non-cross-validated r2 = 0.788, F-value 51.990 and predicted r2 = 0.628. The steric, electrostatic and hydrogen bond donor and hydrogen bond acceptor field contributions were 17.5, 25.3, 27.6 and 29.6%, respectively. The combination of steric, electrostatic, hydrophobic and hydrogen bond donor fields yielded a CoMSIA model with a cross-validated r2 = 0.474 with three components, non-cross-validated r2 = 0.828, F-value 69.674 and predicted r2 = 0.618. The steric, electrostatic, hydrophobic and hydrogen bond donor field contributions were 16.6, 26.3, 29.6 and 27.5%, respectively.
The CoMSIA model with a combination of steric, electrostatic and acceptor fields yielded the highest cross-validated r2 = 0.526 with six components, non-cross-validated r2 = 0.915, F-value 70.215 and the highest predictive r2 of 0.641.
The models generated by various combinations of CoMSIA fields (Table 5) showed moderate to high, internal and external predictions, in which the acceptor fields were dominant over the hydrogen bond donor and hydrophobic fields. The CoMSIA steric and electrostatic contours (not shown) were positioned similarly to those of the CoMFA model as shown in Fig. 2 and 3, hence are not discussed. The hydrogen bond acceptor contour maps of CoMSIA (STDDEV*COEFF) are displayed in Fig. 4.
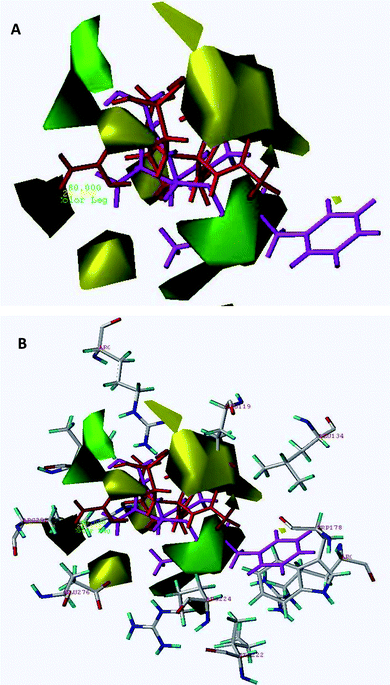 | ||
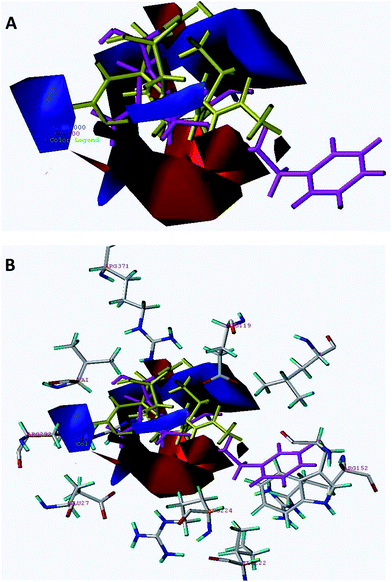
| Fig. 2 The steric contour map of CoMFA. (A) Compound 45 (shown in magenta color) occupies the green colored contour in the region whereas inactive compound 61 (shown in red color) occupies a section of the yellow colored contour in the region. (B) Superimposition of steric contours on A/H1N1 active site. | ||
 | ||
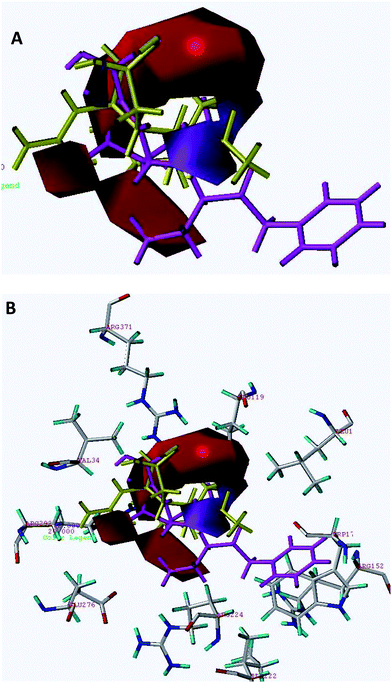
| Fig. 3 (A) The electrostatic contour map of the CoMFA model shown together with the conformations for the most active derivative 45. (B) Superimposition of electrostatic contours on A/H1N1 active site. | ||
 | ||
| Fig. 4 (A) The hydrogen bond acceptor contour maps of the CoMSIA model are shown together with the conformations for the most active derivative 45 (shown in magenta color) and the inactive derivative 61 (shown in yellow color). (B) Superimposition of hydrogen bond acceptor contours on A/H1N1 active site. Cyan polyhedron shows the hydrogen bond favorable region and violet colored polyhedron disfavors the hydrogen bond donor group. | ||
CoMFA versus CoMSIA
In CoMFA, only one model having the two fields, the steric and the electrostatic fields, was possible whereas in CoMSIA seven models were constructed with different combinations of the available fields. The CoMFA model that takes into account steric and electrostatic interactions gave an acceptable rcv2 value of 0.519 and an rpred2 value of 0.605 (Table 4; Alignment III). If identical interactions were assumed for CoMSIA, then a smaller value of rcv2 0.487 and rpred2 0.617 were observed. The best CoMSIA model showed a slightly better rcv2 value i.e. 0.526 and rpred2 value i.e. 0.641 than the best CoMFA model. Hence, as for as predictive ability of the model is considered, the CoMSIA model is better than the CoMFA model.Graphical interpretation of the CoMFA and CoMSIA models
To visualize the information contents of the derived 3D-QSAR models, CoMFA contour maps were generated by interpolating the products between the 3D-QSAR coefficients and their associated standard deviations. The field values generated at each grid point were calculated as the scalar product of the associated QSAR coefficient and the standard deviation of all values in the corresponding column of the data table (STD*COEFF) plotted as the percentage contributions to QSAR equation. The CoMFA steric and electrostatic contour maps shown in Fig. 2 and 3, respectively, are derived from the CoMFA model using docking-based (Alignment-III) active conformation alignment. The 3D contour maps show that the changes in molecular fields are associated with the differences in biological activity. Fig. 2A indicates the CoMFA steric maps obtained from the best model (Alignment-III) using most active compound 45 (shown in magenta color) and least active compound 61 (shown in red color), as reference structures. In this figure, the green contours represent regions of high steric tolerance (80% contribution), while the yellow contours represent regions of low steric tolerance (20% contribution). The green colored regions indicate areas where steric bulk enhances biological activity, while the yellow contours indicate regions where steric bulk is detrimental for biological activity. Blue colored regions show areas where electropositively charged groups enhance activity, while red regions represent regions where electronegatively charged groups improve the activity.Fig. 3A indicates the CoMFA electrostatic contour maps obtained from the best model (Alignment III) using the most active compound 45 (shown in magenta color) and the least active compound 61 (shown in yellow color) as reference structures. In this figure the increase in positive charge is favored in blue regions while the increase in negative charge is favored in red regions.
The steric contour map of CoMFA (Fig. 2A) shows a green contour observed near the 3-substituted phenylpentan-3-yloxy group of cyclohexene of compound 45 and another small green contour to the left side i.e. in the vicinity of carboxylic acid suggesting that steric substituents in these regions may favor activity. Good inhibitory potency of compounds 39 (IC50 = 1.00 nM), 42 (IC50 = 1.00 nM) and 51–54 is due to orientation of bulkier groups towards sterically favored green contours. The disfavored yellow contours surrounding the cyclohexene ring restrict the steric substitution indicating the decrease in biological activity. Compound 61 is the least active compound (shown in red color) since the 4-guanidino group and acetamido-2-[butyl(propyl)amino]-2-oxoethyl group present on the cyclopentane ring are directed completely towards the disfavored yellow region. The same is the case with other compounds having less or moderate activities. Compound 18 (IC50 = 130 µM) is having poor activity since the 5-methylsulfonamido group present on the pyrrolidine ring is directed towards the yellow contour. Compound 16 is having poor activity (IC50 = 96 µM) since 5-acrylamidomethyl and 1-ethyl(isopropyl)carbamoyl groups present on pyrrolidine ring are directed towards the yellow contour. For compound 11 (IC50 = 46 µM) the 2-aminoethyl side chain present on pyrrolidine is completely embedded in the yellow contour present on the upper side i.e. 1-substituted region of the pyrrolidine ring and this could be one of the reasons for compound 11 to have poor biological activity. Compound 2 having an IC50 value of 25 µM is having poor activity since the 3-carboxylic acid group is completely embedded and diethylcarbamoyl group is directed towards the yellow region.
CoMFA electrostatic contour plots displayed a positively charged favored prominent large blue contour near the methyl group of 4-acetamido of compound 45 and two other blue contours, one surrounding the 5-amino group and other in the vicinity of 2nd substituted region of the cyclohexene ring of compound 45. A small red contour is also observed near the carboxyl group, suggesting that the increase in activity may be due to the presence of an electron rich substituent group (compounds 51–53). For compounds 49–53, the C-5 guinidino moiety is completely embedded in the large and prominent blue contour and the carboxyl group is pointing towards the red contour justifying good inhibitory activity for these compounds. The C-4 guanidino group of compound 20 is pointed away from the prominent blue contour and this could be the reason for it having lower biological activity (pIC50 = 4.602).
Two big sized negatively charged favored red contours were observed, one pointing towards the oxygen of the acetamido group and another one in the vicinity of the ethyl group of 3-phenylpropoxy of the cyclohexene group of compound 45 (Fig. 3A). A small red contour was also observed near the oxygen of the 1-ethyl-3-phenylpropoxy side chain present on the cyclohexene of compound 45. Another red contour is present in the vicinity of the carboxylic acid region of compound 45. Close observation for compounds 1–12 suggested that the R1 substitution is pointing away from the blue contour and the carboxyl group away from the red contour, suggesting a lower degree of activity for these compounds. In compound 28, the methyl group of acetamide is pointing towards the red contour and the carbonyl group is placed towards the blue contour, both of which are unfavorable and thus causing the decrease in biological activity.
The CoMSIA steric and electrostatic contours (not shown) were positioned similarly to those of the CoMFA model, hence are not discussed. The hydrogen bond acceptor contour maps of CoMSIA ((STDDEV*COEFF) are displayed in Fig. 4A. In this figure, it is observed that in the highly active template molecule 45 (shown in magenta color), the violet colored contour is in the region of oxygen of the 1-ethyl-3-phenylpropoxy side chain present on the cyclohexene. On the contrary, in the inactive compound 61 (shown in yellow color) there is no hydrogen-bond acceptor group in that region and this could be one of the reasons for poor activity of compound 61. The CoMSIA model generated from steric and electrostatic fields did not vary both in terms of statistical values and positions of contours as compared to its CoMFA model, hence are not discussed.
Superimposition of CoMFA and CoMSIA maps on the active site of A/H1N1
Interaction of Relenza with the active site of A/H1N1 (homology modelled) is shown in Fig. 5. Key interactions of Relenza were observed with the active site through bonding with Glu119, Glu227, Arg292 and Arg371. CoMFA steric and electrostatic contour plots mapped on the active site of A/H1N1 are depicted in Fig. 2B and 3B respectively, with highest active compound 45 (magenta) and lowest active compound 61 (shown in red color in steric and yellow color in electrostatic contour maps). For better perception, only active site residues and nearby residues of the active site i.e. Trp178, Glu119, IIe222, Arg224, Glu227, Glu276, Arg292 and Arg371 were retained for the superimposition of the contour maps on the active site of A/H1N1. The sterically favored big green contour was placed in between Arg224, Arg152 and Leu134 and the other one was placed in the vicinity of Val349, while disfavored yellow contours were observed at the periphery of Glu119, Glu276 and Arg292. The positively charged favored blue contour near the NH2 and acetamido (NHAc) substituent was placed close to Glu276 and Glu119 indicating ionic interactions.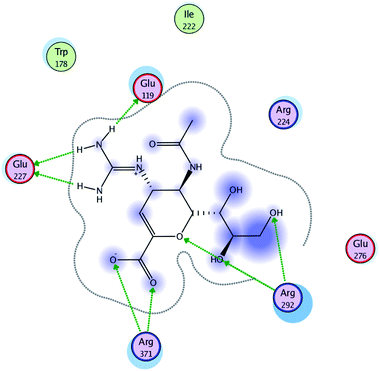 | ||
| Fig. 5 Interaction of Relenza with the active site of A/H1N1. | ||
Fig. 4B displays the CoMSIA acceptor contour maps and their overlapping on the A/H1N1 active site with the most active compound 45 (shown in magenta color) and the least active compound 61 (shown in yellow color). Magenta contour was observed in the vicinity of Arg292 suggesting the hydrogen bond acceptor group in the vicinity would increase the activity. While mapping the contours on the active site an interesting observation was made that aromatic ring present in compound 45 is making hydrophobic interaction with Trp178. This could be one of the reasons for the good activity of compound 45.
Materials and methods
Dataset for CoMFA modeling analysis
As it is well known, the validity of 3D-QSAR analysis is strongly dependent upon the selected training dataset. Therefore, the dataset of compounds with reported bioactivity need to be selected very judiciously. Sixty one molecules selected for the present study were taken from the published work by Wang et al.,13 Chand et al.14,15 and Kim et al.16 The structures of the training and test molecules are given in Table 1. The biological activity used in the present study was expressed as pIC50 = −log IC50 where, IC50 is the molar concentration in micro/nanomoles of the inhibitor producing 50% inhibition of neuraminidase type A.Homology modeling of A/H1N1 and molecular docking
An accurate 3D structure of the target enzyme is important for molecular docking and rational development of novel potential inhibitors. Since the three dimensional structure of A/H1N1 neuraminidase currently is not known, the homology model of A/H1N1 neuraminidase bound with Relenza developed by our group6 by using H5N1 as the starting point was used for docking studies. Molecular docking was carried out by using the Glide module of Schrodinger package according to the previously reported protocol.17,183D QSAR CoMFA and CoMSIA modeling
Molecular modeling
Molecular modeling, CoMFA and CoMSIA analysis were performed using SYBYL 7.0 software19 running on Silicon Graphics Fuel workstation. The 3D structures of molecules were built using the ‘Sketch Molecule’ function within SYBYL. Initial optimization of the structures was carried out using Tripos force fields with MMFF94 charges and repeated minimization was performed using steepest descent and conjugated gradient methods till the root-mean-square (rms) deviation of 0.001 kcal mol−1 was achieved. Conformational energies were computed with electrostatic terms; the lowest energy structures finally minimized were used in superimpositions and docking studies. The partial atomic charges required for the electrostatic interactions were computed by a semi-empirical molecular orbital method using MOPAC20 with AM1 Hamiltonian.21Molecular alignment rules for CoMFA modeling
In 3D QSAR, the determination of the bioactive conformer and molecular alignment of the compounds is an important step. Molecular superposition plays a decisive role in CoMFA analysis, since the relative interaction energies depend strongly on relative molecular positions. Even though the automatic alignment rules are widely practiced in the CoMFA approach with high performance, the manual alignment method is a competent alternative for yielding high predictive models as well.22 Hence we thought logical to generate low energy conformer of the template molecule and study the outcome of the result on the quality of the CoMFA modeling by using different alignment methods. Two different methods were used for defining alignment rules in the CoMFA modeling study as described below:(a) Alignment II: docked conformations were aligned on one another and the same were used for the CoMFA model development.
(b) Alignment III: the lowest energy conformer of compound (45) obtained through dynamics using the simulated annealing technique (explained in Method 1) was submitted for docking and this docked conformer was used for alignment. After docking all the conformations in the active site, all the compounds were aligned on the above obtained conformation of compound (45) as per the procedure adopted in Method 1.
CoMFA interaction energy calculation
The steric and electrostatic fields were calculated at each lattice intersection of a regularly spaced grid of 2.0 Å in all the three dimensions within a defined region. The grid pattern was generated automatically by the SYBYL/CoMFA routine. The van der Waals potentials and coulombic terms representing the steric and electrostatic fields, respectively, were calculated using standard Tripos force fields. A distance-dependent dielectric constant of 1.00 was used. An sp3carbon atom with +1.0 charge was used as a probe atom. The steric and electrostatic fields were truncated at +30.0 kcal mol−1.CoMSIA interaction energy calculation
In CoMSIA interaction energy calculations, the steric, electrostatic, hydrophobic, hydrogen bond donor and hydrogen bond acceptor potential fields were calculated at each lattice intersection of a regularly spaced grid of 2.0 Å. A probe atom with radius 1.0 Å and +1.0 charge with hydrophobicity of +1.0 and hydrogen bond donor and hydrogen bond acceptor properties of +1.0 were used to calculate steric, electrostatic and hydrophobic fields. Contributions from these descriptors were truncated at 0.3 kcal mol−1.Partial least square (PLS) analysis
The PLS approach,23,24 which is an extension of the multiple regression analysis, was used to derive the 3D-QSAR models, in which the CoMFA descriptors were used as independent variables, and the experimental pIC50 values were used as dependent variables. The cross-validation25 analysis was performed for selecting which model, among several ones with different levels of complexity, is most likely to have predictive values. The correctness intensity of the cross-validation process is controlled by selecting a group of ten compounds. The cross-validated r2 that resulted in the optimum number of components and the lowest standard error of prediction were considered for further analysis. Equal weights were assigned to steric and electrostatic fields using the CoMFA_STD scaling option. To speed up the analysis and reduce noise, a minimum filter value (σ) of 2.0 kcal mol−1 was used. Final analysis was performed to calculate conventional r2 using the optimum number of components. The entire cross-validated results were analyzed by considering the fact that a value of rcv2 above 0.3 indicated the probability of chance correlation to be less than 5%.26 The q2 (cross-validated r2), SPRESS (cross-validated standard error of prediction), r2 (non-cross-validated r2), F values and standard error of the estimate value were computed according to the definitions in SYBYL 7.0 package.Conclusions
In the present study, sixty one neuraminidase type A inhibitors were selected for the development of 3D-QSAR CoMFA and CoMSIA models. 3D-QSAR CoMFA predictive models were established for the neuraminidase type A inhibitors by using two different molecular alignment strategies one with atom and centroid/atom based alignment and second with docking conformer based alignment. Homology modelled conformation of A/H1N1 structure was used for docking purpose. The model developed by considering the docked structure-based conformers having atom and centroid/atom fitting was found to perform better than the one developed by atom and centroid/atom fit based, with excellent cross-validated r2 and predictive r2 values. The steric and electrostatic field contributions revealed relatively higher contributions of steric fields. The CoMFA model generated using docked conformer-based alignment having atom and centroid/atom based fitting served for alignment for CoMSIA studies also. The CoMSIA model with a combination of steric, electrostatic and acceptor fields yielded the highest cross-validated r2. As far as predictive ability is considered, the CoMSIA model showed better predictive ability than the CoMFA model. CoMFA steric, electrostatic and CoMSIA acceptor contour maps were mapped in the active site of homology modelled A/H1N1 conformations. The important outcome of this study indicates that sterically favored bulkier groups should be maintained in the vicinity of Arg224, Arg152, Leu134 and Val349, while bulky groups should be avoided in the periphery of Glu119, Glu276 and Arg292 for better activity. By keeping the hydrogen bond acceptor group in the vicinity of Arg292 would also increase the activity. Overall, the present 3D-QSAR study investigates the indispensable structural features of different chemical classes of molecules which can be exploited for the structural modifications of these lead molecules in order to achieve improved neuraminidase inhibitory activity.References
- Y. S. Babu, P. Chand, S. Bantia, P. Kotian, A. Dehghani, Y. El-Kattan, T. H. Lin, T. L. Hutchison, A. J. Elliott, C. D. Parker, S. L. Ananth, L. L. Horn, G. W. Laver and J. A. Montgomery, J. Med. Chem., 2000, 43, 3482–3486 CrossRef CAS.
- J. L. McKimm-Breschkin, Antiviral Res., 2000, 47, 1–17 Search PubMed.
- B. McKay, Wall St. J., March 2, 2010 Search PubMed.
- G. N. T. Neumann and Y. Kawaoka, Nature, 2009, 459, 931–939 Search PubMed.
- Pandemic (H1N1) 2009 briefing note 1, http://www.who.int/csr/disease/swineflu/notes/h1n1_antiviral_resistance_20090708/en/.
- L. Le, E. Lee, K. Schulten and T. N. Truong, PLoS Curr. Influenza, 2009, RRN1015 Search PubMed.
- H. T. Nguyen, L. Le and T. N. Truong, PLoS Curr. Influenza, 2009, RRN1030 Search PubMed.
- P. R. Murumkar, R. Giridhar and M. R. Yadav, Chem. Biol. Drug Des., 2008, 71, 363–373 Search PubMed.
- P. R. Murumkar, S. D. Gupta, V. P. Zambre, R. Giridhar and M. R. Yadav, Chem. Biol. Drug Des., 2009, 73, 97–107 Search PubMed.
- P. R. Murumkar, V. P. Zambre and M. R. Yadav, J. Comput.-Aided Mol. Des., 2010, 24, 143–156 Search PubMed.
- V. P. Zambre, P. R. Murumkar, R. Giridhar and M. R. Yadav, J. Chem. Inf. Model., 2009, 49, 1298–1311 Search PubMed.
- V. P. Zambre, P. R. Murumkar, R. Giridhar and M. R. Yadav, J. Mol. Graphics Modell., 2010, 29, 229–239 Search PubMed.
- G. T. Wang, Y. Chen, S. Wang, R. Gentles, T. Sowin, W. Kati, S. Muchmore, V. Giranda, K. Stewart, H. Sham, D. Kempf and W. G. Laver, J. Med. Chem., 2001, 44, 1192–1201 Search PubMed.
- P. Chand, Y. S. Babu, S. Bantia, S. Rowland, A. Dehghani, P. L. Kotian, T. L. Hutchison, S. Ali, W. Brouillette, Y. El-Kattan and T. H. Lin, J. Med. Chem., 2004, 47, 1919–1929 Search PubMed.
- P. Chand, P. L. Kotian, A. Dehghani, Y. El-Kattan, T. H. Lin, T. L. Hutchison, Y. S. Babu, S. Bantia, A. J. Elliott and J. A. Montgomery, J. Med. Chem., 2001, 44, 4379–4392 Search PubMed.
- C. U. Kim, W. Lew, M. A. Williams, H. Wu, L. Zhang, X. Chen, P. A. Escarpe, D. B. Mendel, W. G. Laver and R. C. Stevens, J. Med. Chem., 1998, 41, 2451–2460 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelly, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1339–1349 Search PubMed.
- M. Clark, R. D. Cramer and N. J. Van Opdenbosh, J. Comput. Chem., 1989, 982–1012 Search PubMed.
- SYBYL Molecular Modeling System, Version 7.0, 2003 Search PubMed.
- J. J. Stewart, J. Comput.-Aided Mol. Des., 1990, 4, 1–105 Search PubMed.
- M. J. Dewar, E. F. Healy and J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 Search PubMed.
- L. Zhang, K.-C. Tsai, L. Du, H. Fang, M. Li and W. Xu, Curr. Med. Chem., 2011, 18, 923–930 Search PubMed.
- S. R. A. Wold, H. Wold and W. J. I. Dunn, SIAM J. Sci. Stat. Comput., 1984, 5, 735–743 Search PubMed.
- M. Clark and R. D. Cramer, Quant. Struct.–Act. Relat., 1993, 12, 137–145 Search PubMed.
- B. L. Podlogar and D. M. Fergusson, Drug Des. Discovery, 2000, 17, 4–12 Search PubMed.
- M. Clark, R. D. Cramer, D. Jones, D. E. Patterson and P. Simeroth, Tetrahedron Comput. Methodol., 1990, 3, 47–59 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |