Triazole-substituted N-hydroxyindol-2-carboxylates as inhibitors of isoform 5 of human lactate dehydrogenase (hLDH5)†
Carlotta
Granchi
a,
Sarabindu
Roy
a,
Claudia
Del Fiandra
a,
Tiziano
Tuccinardi
a,
Mario
Lanza
b,
Laura
Betti
a,
Gino
Giannaccini
b,
Antonio
Lucacchini
b,
Adriano
Martinelli
a,
Marco
Macchia
a and
Filippo
Minutolo
*a
aDipartimento di Scienze Farmaceutiche, Università di Pisa, Via Bonanno 6, 56126, Pisa, Italy. E-mail: filippo.minutolo@farm.unipi.it; Fax: +39 050 2219605; Tel: +39 050 2219557
bDipartimento di Psichiatria, Neurobiologia, Farmacologia e Biotecnologie, Università di Pisa, Via Bonanno 6, 56126, Pisa, Italy
First published on 17th May 2011
Abstract
hLDH5 plays a key role in the glycolytic metabolic switch found in invasive tumours and is now considered as a promising therapeutic target in cancer. Here we report a series of new hLDH5-inhibitors based on triazole-containing N-hydroxyindol-2-carboxylates, whose synthesis includes the highly reliable and simple “click chemistry” reaction.
Lactate dehydrogenase (LDH) is a widely diffused 2-hydroxyacid oxidoreductase, which promotes the inter-transformation of pyruvate and lactate by using a nicotinamide adenine dinucleotide cofactor (NADH/NAD+). Various isoforms of LDH are presently being considered as promising targets for a range of pathologies, such as cancer and malaria.1
Human isoforms of LDH are tetrameric structures composed of ∼35 kDa protein subunits referred to as LDH-A, LDH-B and LDH-C. Various combinations of the A and B subunits lead to the formation of five isoforms (hLDH1-5) consisting in either homotetramers, such as hLDH1 (LDH-B4) and hLDH5 (LDH-A4), or heterotetramers made of the other three possible combinations of A and B subunits (hLDH2–4).2 The respective localizations of the homotetramers are quite distinct: hLDH1 is the main isoform present in the heart and is mostly concentrated in the mitochondiral matrix, whereas hLDH5 largely prevails in the skeletal muscle (equally present in mitochondria and cytosol) and liver (more abundant in mitochondria).3 In contrast, the C subunit seems to take part only as a homotetrameric enzyme, hLDHX (LDH-C4), which plays a role in male fertility and, therefore, it appears totally inactive in females.4
Over the past few years, hLDH5 has been found to be overexpressed in highly invasive and hypoxic carcinomas,5 and gene expression of the LDH-A subunit was demonstrated to be inversely correlated to the oxygen content in cancer cell culture media.6hLDH5-overexpression was associated to the peculiar metabolic switch occurring in invasive tumours, also known as the “Warburg effect”.7 This metabolic adaptation consists in an increased glycolysis producing energy and anabolites from glucose even under hypoxic conditions, which are widely diffused in radio-/chemo-resistant metastatic cancer.8 Since healthy tissues mostly rely on oxidative phosphorylation for energy production, at least under normal conditions, the glycolytic character of tumour cells has emerged as a possible “cancer's Achilles' heel”.9 In particular, among the several enzymes involved in glycolysis, hLDH5 is currently being considered as one of the most suitable check-points, where a therapeutic interference (by means of enzyme inhibition) should be able to efficiently counteract cancer invasion, without seriously compromising the regular carbohydrate metabolism occurring in normal cells.1 In fact, gene repression of the production of the LDH-A subunit in cancer cells by shRNA seriously compromised the tumourigenicity of these LDH-A-deficient cells under hypoxia.10 Moreover, studies on naturally occurring knock-out human “models”, represented by people affected by hereditary LDH-A deficiency, show that this condition does not cause any symptoms under ordinary circumstances (muscle damage and consequent myoglobinuria are observed only after intense anaerobic exercise).11 This evidence would support the hypothesis that hLDH5 is a safe target for anticancer therapy.
So far, only very few successful attempts to selectively inhibit hLDH5 by means of small organic “drug-like” compounds with the purpose of blocking glycolysis in cancer cells have been reported in the literature.1 An analysis of the hLDH5 active site reveals that it is placed in a rather deep position within the protein and accessibility to it is narrow.2 Furthermore, the enzyme cavity is rich in cationic residues (arginines). Therefore, prospective inhibitors possessing deprotonatable groups, such as COOH, are generally preferred for an efficient interaction with the active site. All these features have made the production of highly potent hLDH5-inhibitors (Ki or IC50 values below the micromolar range) particularly challenging. One of the latest reports includes an 8-deoxyhemigossylic acid derivative (FX11), an efficient inhibitor of hLDH5 (Ki = 8 μM), which proved to block tumour progression both in vitro and in vivo.12 Another very interesting approach is represented by the development of “bifunctional” hLDH5-inhibitors, where pyruvate-like alkanoic acid fragments, mimicking the natural enzyme substrate, were linked to an adenosine-like bis(indolyl)maleimide portion, which was intended to occupy part of the cofactor binding pocket. Some of these compounds showed IC50 values in the low micromolar range (14.8–35.9 μM).13
We recently reported on a new class of hLDH5-inhibitors, consisting of variously substituted N-hydroxyindol-2-carboxylates (NHIs).14 Some of these compounds, bearing a phenyl group in position 5 or 6, proved to be efficient inhibitors of hLDH5 (Ki's ranging from 4.7 μM to 35.4 μM), with a high degree of isoform selectivity, since their inhibition of hLDH1 (exclusively composed of the LDH-B subunit) was always very low. Most importantly, enzyme kinetic experiments showed that these NHI-based inhibitors are competitive with respect to both NADH and pyruvate. This supports our molecular modelling analysis which indicates that these compounds contemporarily occupy the substrate-binding pocket with their “OH/COOH” polar terminal, and the cofactor-binding pocket with their aromatic portions. Preliminary cell-based assays revealed that these NHIs are able to reduce lactate production in cancer cells and they possess a strong antiproliferative activity, which is particularly pronounced under hypoxic conditions.14 Due to the initial success obtained within the NHI class, we then started to consider possible molecular variations that could expand the series of these selective hLDH5-inhibitors.
Herein, we report the development of an investigative series of NHI derivatives containing 1,2,3-triazole portions in their structures. Triazoles are often introduced into potentially bioactive molecules, including the above-mentioned “bifunctional” hLDH5-inhibitors,13 mostly because of their readily synthetic accessibility by a modification of the Huisgen reaction, consisting of a simple Cu(I)-catalysed 1,3-dipolar cycloaddition between azides and alkynes, also known as “click-reaction”.15 The ease and efficiency of this reaction makes it a very suitable and versatile synthetic tool for the fast obtainment of vast arrays of bioactive compounds. Nevertheless, the significant dipole moment of the triazole core may, in some cases, considerably affect the interaction of the resulting molecules with their biological target.16 Therefore, our first concern was to verify if the presence of triazole portions on the NHI scaffold could be tolerated in terms of maintenance of hLDH5-inhibitory efficacy. To accomplish this, we based our molecular design on modelling studies performed on our first series of NHIs docked into the LDH-A active site, constituting the sole subunit present in the tetrameric functional form of hLDH5. These studies revealed that there is a substantial space for chemical expansion around the 6-member ring of the NHI scaffold, corresponding to the NADH-binding site of the enzyme, placed in proximity of the enzyme entrance channel, whereas the polar terminal including the hydroxyl/carboxylate motif is deeply buried in the small pyruvate-binding pocket (Fig. 1). Therefore, we replaced the phenyl substituent of our original NHIs14 with variously substituted triazole moieties, thus generating a new exploratory series of triazole–NHIs (1, Fig. 1).
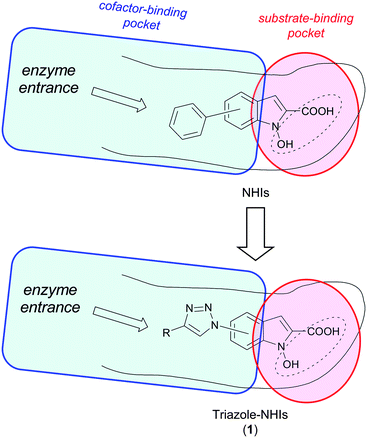 | ||
| Fig. 1 Simplified models of occupancy of NHIs within LDH-A active site and structural derivation of new triazole–NHIs (1). | ||
For this purpose, we synthesized NHI derivatives containing the triazole-substituent in the 4- (1a–d), 5- (1e,f) and 6-position (1g) of the NHI scaffold, as well as a dimeric analogue (2) (Fig. 2). In addition to the indications derived from modelling studies, the choice of the compounds herein reported was mainly dictated by the synthetic accessibility and the chemical stability of the resulting target molecules.
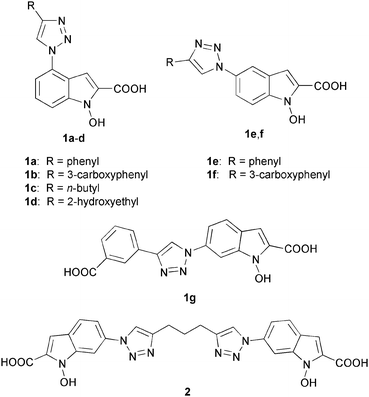 | ||
| Fig. 2 Structures of newly synthesized triazole-substituted N-hydroxyindole-2-carboxylic acids (1a–g and 2). | ||
The synthesis of compounds 1a–g (Scheme 1) starts with the copper-catalyzed cycloaddition of aryl-azides 3 with the appropriate terminal alkynes (4), leading to the formation of triazole-substituted nitrotoluene derivatives 5. Subsequent reaction with sodium hydride and dimethyl oxalate led to the formation of α-ketoesters 6.17 Then, the outcome of the reductive cyclization step proved to be highly dependent on the substrate. In fact, the concurrent formation of the over-reduced NH–indole side product became significant in some cases, as we had previously verified for other types of substrates.14 For most ketoesters (6a–f) the best reaction conditions turned out to be the employment of stannous chloride dihydrate in the presence of 4 Å molecular sieves in anhydrous DMF,17 which promoted the formation of the desired N-hydroxyindole cycle in low-to-moderate isolated yields. However, when these conditions were applied to precursor 6g, over-reduction to the indole side product became highly predominant. Therefore, we changed the reaction conditions and used lead/triethylformate (TEAF) in refluxing methanol.18 This way we were able to selectively obtain N-hydroxyindole 7g. Final hydrolysis with LiOH generated the desired NHIs derivatives 1a–g.
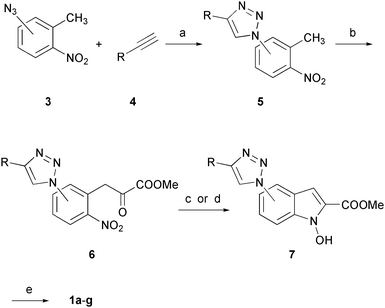 | ||
Scheme 1
Reagents and conditions: (a) CuSO4·5H2O, sodium ascorbate, t-BuOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 80 °C, 24 h, 35–97%, or μW, 15 min, 73–91%; (b) (COOMe)2, NaH, DMF, 0 °C to RT, 4–16 h, 40–81%; (c) SnCl2·2H2O, DME, 4 Å MS, 0 °C, 2–12 h, 35–63% (compounds 7a–f); (d) Pb, HCO2HNEt3, MeOH, reflux, 18 h, 40% (compound 7g); (e) aqueous 2N LiOH, THF/MeOH (1:1), RT, 1–24 h, 89–98%. :1), 80 °C, 24 h, 35–97%, or μW, 15 min, 73–91%; (b) (COOMe)2, NaH, DMF, 0 °C to RT, 4–16 h, 40–81%; (c) SnCl2·2H2O, DME, 4 Å MS, 0 °C, 2–12 h, 35–63% (compounds 7a–f); (d) Pb, HCO2HNEt3, MeOH, reflux, 18 h, 40% (compound 7g); (e) aqueous 2N LiOH, THF/MeOH (1:1), RT, 1–24 h, 89–98%. | ||
Dimeric NHI 2 was synthesized as shown in Scheme 2. In this case, we applied the “click reaction” to 1,6-heptadiyne (8), which reacted with two molecules of azide 3c, giving bis-triazole derivative 9. Subsequent NaH-promoted double condensation of 9 with dimethyloxalate afforded bis(α-ketoester) 10. Reductive cyclization of 10 required a further optimization, since both stannous chloride and lead/TEAF conditions gave rise to complex reaction mixtures composed of cyclized products where one or both of the new cycles were over-reduced to N–H indoles, together with the desired product. Moreover, product separation and purification from these mixtures proved to be particularly difficult. In this case, we found that reaction of 10 with sodium hypophosphite in the presence of palladium 10% on charcoal, previously reported to selectively promote the partial reduction of nitro-groups to hydroxylamines,19 produced the formation of bis(N-hydroxyindole) 11, without significant production of over-reduced side-products. This way, after final hydrolysis with LiOH, we were able to obtain bis(NHI) 2.
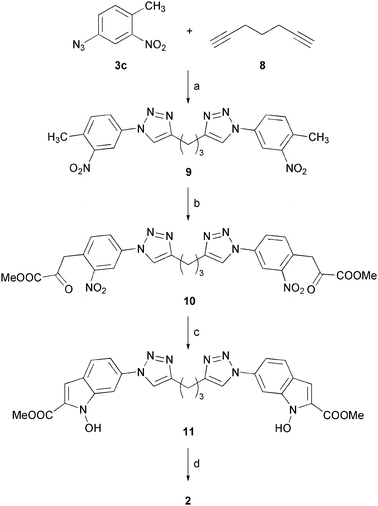 | ||
| Scheme 2
Reagents and conditions: (a) CuSO4·5H2O, sodium ascorbate, t-BuOH/H2O (1:1), RT, 48 h, 75%; (b) (COOMe)2, NaH, DMF, 0 °C to RT, 16 h, 68%; (c) H2PO2Na·H2O, Pd–C, H2O/THF (1:1), RT to 40 °C, 13 h, 34%; (d) aqueous 2 N LiOH, THF/MeOH (1:1), RT, 12 h, 89%. | ||
The LDH inhibitory activities of compounds 1a–g and 2 were measured by standard enzyme kinetic experiments on purified human enzyme isoforms hLDH5 (or LDH-A4) and hLDH1 (or LDH-B4). In a preliminary screening, we verified the percent inhibition of hLDH5 relative to control at a compound concentration of 125 μM in the presence of 25 μM NADH. Enzyme activity was determined by the measurement of the absorbance decrease at 340 nm, due to the consumption of NADH, and the percentage inhibition relative to control was calculated. We decided to further evaluate only compounds displaying a > 50% inhibition at that concentration. From this screening, compounds 1c–f did not show appreciable levels of inhibition (<15% in all cases), whereas the other compounds (inhibition %, see Table 1), 1a,b,g and 2, were selected for a complete enzyme kinetic analysis to verify their type of inhibition versus both the cofactor and the substrate, as well as their isoform selectivity (hLDH5vs. hLDH1). For this purpose, we first evaluated the apparent Michaelis–Menten constant (Km) values of cofactor (NADH) and substrate (pyruvate) for hLDH5, measured from Lineweaver–Burk plots. Then, we measured the apparent Km′ values in the presence of the above-mentioned inhibitors (concentration range = 25–100 μM). From the values of Km′ so obtained, Ki values for each inhibitor were determined by double-reciprocal Lineweaver–Burk plots and their values are reported in Table 1.
| Cmpd | hLDH5 | hLDH1 | |||
|---|---|---|---|---|---|
| % inhib. a | K i/μM,b [NADH]c | K i/μM,b [Pyr]d | % inhib. a | K i/μM,b [NADH]c | |
| a Inhibition % was measured at 125 μM of each compound, 25 μM of NADH and 2 mM of sodium pyruvate. b Average of two or more measurements. The error in these values is within ±30% of the average. c Saturating concentration (2.0 mM) of sodium pyruvate and competitive increasing concentrations (12.5 ÷ 150 μM) of NADH. d Saturating concentration (200 μM) of NADH and competitive increasing concentrations (25 μM ÷ 1.0 mM) of sodium pyruvate. e Not determinable. | |||||
| 1a | 73% | 28 | 44 | <3% | nde |
| 1b | 60% | 57 | 86 | 60% | 89 |
| 1g | 65% | 50 | 45 | <3% | nde |
| 2 | 83% | 20 | 37 | 32% | nde |
First of all, it is worth mentioning that hLDH5-kinetic experiments conducted with all of these compounds showed that variations in the concentrations of inhibitors only caused changes in the Km′ values, whereas Vmax was not affected. This demonstrates that 1a,b,g and 2 are competitive inhibitors of hLDH5, and they manifest a competitive behaviour vs. both the cofactor (NADH) and the substrate (pyruvate), similarly to their previously reported analogues.14 Furthermore, these compounds displayed a preferential inhibition of isoform 5, with the single exception of compound 1b, which proved to be an indiscriminate inhibitor of both isoforms. The Ki values obtained with hLDH5 revealed that compounds 1a and 2 are the most efficient inhibitors of this enzyme isoform, reaching relatively low micromolar levels in the competition assays vs.NADH (28 μM for 1a and 20 μM for 2). Compound 1a displayed a high selectivity for hLDH5 because its inhibition of the other isoform hLDH1 was negligible. A sharp preference for hLDH5 was also found with compound 2, although in this case a residual 32% inhibition of hLDH1 at 125 μM was observed. These inhibition data indicate that the presence of a phenyl-substituted triazole group in position 4 of the NHI scaffold (1a), as well as the dimeric motif generated by linking two NHI moieties at their 6 positions with a 1,3-bis(triazole)-propane chain (2), are nicely tolerated by the enzyme.
We wanted to understand the way these molecules interact with hLDH5. For this purpose, we conducted molecular modelling studies of the newly synthesized compounds, which were docked into the active site of the LDH-A subunit. As shown in Fig. 3A, in agreement with our previously published results,14 the carboxylic group of compound 1a shows strong polar interactions with R169 and T248 and the N-hydroxy group displays a H-bond with the backbone nitrogen atom of T248, as well as with a water molecule that mediates the interaction with H193. The triazole ring in position 4 of the NHI scaffold of compound 1a does not seem to play an active role in the formation of significant attractive interactions with the protein, but it is fundamental for the good orientation of the phenyl substituent that shows an efficient π–π interaction with the phenolic ring of Y239.
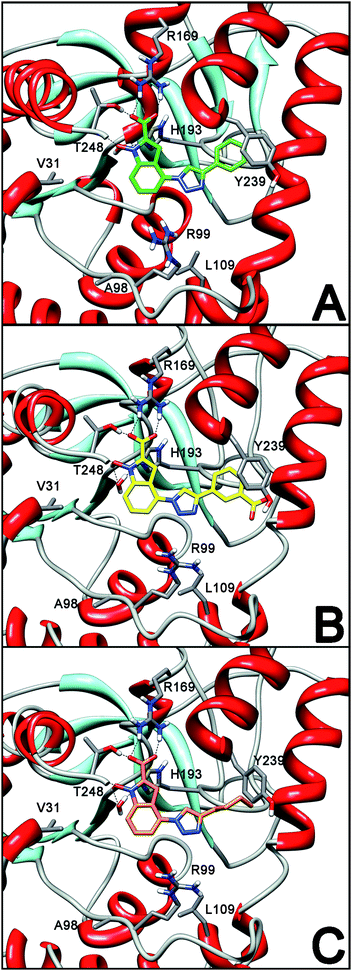 | ||
| Fig. 3 Docking analysis into LDH-A subunit of (A) 1a (green); (B) 1b (yellow); (C) 1c (orange). | ||
The addition of a carboxylic group in position 3 of the phenyl substituent (compound 1b) causes a slight decrease in biological activity and this is confirmed by the modelling results showing that this group does not participate in any important interaction with the enzyme (Fig. 3B), whereas the phenyl ring is still able to form a stable π–π interaction with Y239. On the other hand, the replacement of the 4′-phenyl ring of compounds 1a,b with alkyl chains, either hydrophobic (1-butyl-, 1c) or hydrophilic (2-hydroxyethyl-, 1d), causes a dramatic drop in their inhibitory activities, due to the loss of the above-mentioned π–π interaction, together with the absence of significant alternative interactions with the active site involving the triazole ring and its alkyl substituents. This situation is shown in Fig. 3C by the docking results obtained with compound 1c as a representative example.
The presence of a substituted triazole ring in position 5 determines the loss of LDH activity. In fact, as shown in Fig. 4, the triazole ring of compound 1e does not seem to interact with any residues and the phenyl substituent shows only a putative weak lipophilic interaction with L109.
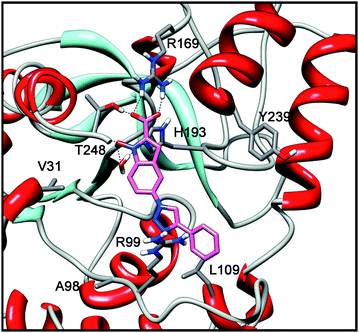 | ||
| Fig. 4 Docking analysis of 1e into LDH-A subunit. | ||
The shift of the substituted triazole ring from position 5 to position 6 determines a high improvement of the inhibitory activity. In fact, the docking analysis of compound 1g coherently highlights that, in addition to the previously described interactions of the NHI scaffold with the protein, the triazole ring in position 6 is able to form a H-bond with R99. Moreover, the aryl substituent efficiently interacts with lipophilic residues V31 and A98 (Fig. 5).
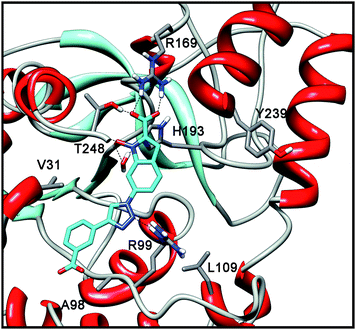 | ||
| Fig. 5 Docking analysis of 1g into LDH-A subunit. | ||
A similar binding pose was found for dimeric derivative 2, where the two NHI moieties are bound through their triazole substituents in position 6 (Fig. 6). In this case, one NHI scaffold is buried inside the enzyme active site and forms the same strong polar interactions described above for its NHI analogues with R169, H193 and T248. The triazole ring bound to the 6 position of this first NHI portion confirms to be placed in an optimal position for the formation of a stable H-bond with R99 (Fig. 6A). Furthermore, the second NHI portion of compound 2 is placed outside the enzyme cavity and it lays into an external groove of the protein, where the adenosine portion of NADH is normally hosted (Fig. 6B). In this position, this group is able to actively participate in the binding process. In fact, a H-bond between the N-hydroxy group of this “outer” NHI scaffold and the backbone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen of residue R99 is observed (Fig. 6A), thus explaining the good inhibitory activity found with compound 2. Of course, any conclusion deriving from molecular modelling should be further validated by X-ray studies, and efforts to obtain crystal structures of the complexes of the LDH-A subunit with our inhibitors are currently underway.
O oxygen of residue R99 is observed (Fig. 6A), thus explaining the good inhibitory activity found with compound 2. Of course, any conclusion deriving from molecular modelling should be further validated by X-ray studies, and efforts to obtain crystal structures of the complexes of the LDH-A subunit with our inhibitors are currently underway.
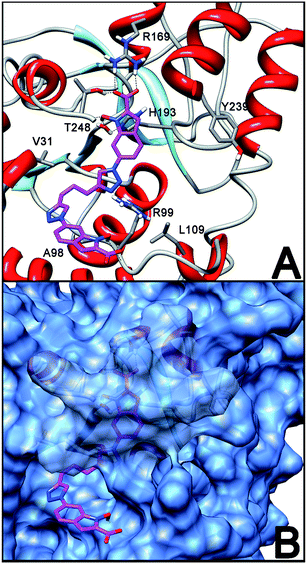 | ||
| Fig. 6 Putative binding pose of 2 into LDH-A subunit (A) and its disposition relative to the outer protein surface (B). | ||
Overall, these results obtained by molecular modelling methods support the experimental good inhibitory activities of compounds 1a,b,g and 2, although their potencies are not superior to those previously found with more hydrophobic NHI-based hLDH5-inhibitors that do not contain triazole substituents.14 A factor which may negatively affect the presence of a polar triazole portion (or even a general extra-polar group) is constituted by the energy of desolvation, which consists of an energy cost occurring during the formal “stripping” of water from the ligand during the binding process. In fact, if the polar portions of the inhibitors are not actively involved in the constitution of strong hydrophilic interactions with the target protein, their binding with the enzyme active site and, therefore, their inhibitory ability may be penalized by this energetic factor.20 Nevertheless, the importance of the findings herein reported consists in the fact that “synthetically attractive” triazole portions, when appropriately positioned in the NHI scaffold, are adequately tolerated by the enzyme active site and, even more, may actively contribute to the binding process.
Conclusions
We have developed an exploratory series of NHIs that are characterized by the presence of triazole portions, as inhibitors of hLDH5. The synthesis of these inhibitors involves an application of the highly reliable and versatile triazole reaction formation, also known as “click reaction”. Experimental and theoretical results show that insertion of triazole rings in the structure of these inhibitors may be highly tolerated by the enzyme active site. Therefore, more extensive series of triazole-based hLDH5-inhibitors may be easily developed in the future by this method, thus increasing the chances of finding new analogues displaying more potent enzyme inhibition levels. Further biological testing with a panel of cancer cell-lines is currently underway and will be reported in due course.Acknowledgements
We thank the European Community (FP7-PEOPLE-IIF-2008-235016; “NOXYCANCERSTARV”) for financial support (FM, SR), Siena Biotech S. p. A. for a research contribution (FM), and the Italian Ministry for University and Research (MIUR) for a PhD fellowship (CG). Thanks to Dr Giorgio Placanica and Dr Caterina Orlando for technical assistance in the analysis of the chemical products.Notes and references
- C. Granchi, S. Bertini, M. Macchia and F. Minutolo, Curr. Med. Chem., 2010, 17, 672 Search PubMed.
- J. A. Read, V. J. Winter, C. M. Eszes, R. B. Sessions and R. L. Brady, Proteins, 2001, 43, 175 CrossRef CAS.
- G. A. Brooks, H. Dubouchaud, M. Brown, J. P. Sicurello and C. E. Butz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1129 Search PubMed.
- E. Goldberg, Exp. Clin. Immunogenet., 1985, 2, 120 Search PubMed.
- M. I. Koukorakis, A. Giatromanolaki, C. Simopoulos, A. Polychronidis and E. Sivridis, Clin. Exp. Metastasis, 2005, 22, 25 Search PubMed; M. I. Koukorakis, M. Pitiakoudis, A. Giatromanolaki, A. Tsarouha, A. Polychronidis, E. Sivridis and C. Simopoulos, Cancer Sci., 2006, 97, 1056 Search PubMed.
- B. S. Sørensen, J. Hao, J. Overgaard, H. Vorum, B. Honoré, J. Alsner and M. R. Horsman, Radiother. Oncol., 2005, 76, 187 Search PubMed; B. S. Sørensen, J. Alsner, J. Overgaard and M. R. Horsman, Radiother. Oncol., 2007, 83, 362 Search PubMed.
- O. Warburg, Science, 1956, 123, 309 CrossRef CAS.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891 CrossRef CAS; M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029 CrossRef CAS.
- G. Kroemer and J. Pouyssegur, Cancer Cell, 2008, 13, 472 CrossRef CAS.
- H. Shim, C. Dolde, B. C. Lewis, C.-S. Wu, G. Dang, R. A. Jungmann, R. Dalla-Favera and C. V. Dang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6658 Search PubMed; V. R. Fantin, J. St-Pierre and P. Leder, Cancer Cell, 2006, 9, 425 CrossRef CAS.
- T. Kanno, K. Sudo, M. Maekawa, Y. Nishimura, M. Ukita and K. Fukutake, Clin. Chim. Acta, 1988, 173, 89 Search PubMed.
- A. Le, C. R. Cooper, A. M. Gouw, R. Dinavahi, A. Maitra, L. M. Deck, R. E. Royer, D. L. Vander Jagt, G. L. Semenza and C. V. Dang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2037 CrossRef CAS.
- A. D. Moorhouse, C. Spiteri, P. Sharma, M. Zloh and J. E. Moses, Chem. Commun., 2011, 47, 230 RSC.
- C. Granchi, S. Roy, C. Giacomelli, M. Macchia, T. Tuccinardi, A. Martinelli, M. Lanza, L. Betti, G. Giannaccini, A. Lucacchini, N. Funel, L. G. León, E. Giovannetti, G. J. Peters, R. Palchaudhuri, E. C. Calvaresi, P. J. Hergenrother and F. Minutolo, J. Med. Chem., 2011, 54, 1599 Search PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS; G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS.
- M. Begtrup, C. J. Nielsen, L. Nygaard, S. Samdal, C. E. Sjøgren and G. O. Sørensen, Acta Chem. Scand., Ser. A, 1988, 42, 500.
- K. C. Nicolaou, A. A. Estrada, G. C. Freestone, S. H. Lee and X. Alvarez-Mico, Tetrahedron, 2007, 63, 6088 CrossRef CAS.
- C. Cesario and M. J. Miller, Org. Lett., 2009, 11, 1293 CrossRef CAS.
- I. D. Entwistle, T. Gilkerson, R. A. W. Johnstone and R. P. Telford, Tetrahedron, 1978, 34, 213 CrossRef CAS.
- A. González, M. Murcia, B. Benhamú, M. Campillo, M.-L. López-Rodríguez and L. Pardo, Med. Chem. Commun., 2011, 2, 160 RSC; Y. Takamatsu and A. Itai, Proteins, 1998, 33, 62 Search PubMed; G. E. Kellogg, J. C. Burnett and D. J. Abraham, J. Comput. Aided Mol. Des., 2001, 15, 381 Search PubMed; A. Morton, W. A. Baase and B. W. Matthews, Biochemistry, 1995, 34, 8564 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, molecular modelling details and characterization data for all new compounds. See DOI: 10.1039/c1md00071c |
| This journal is © The Royal Society of Chemistry 2011 |