Identification of target family directed bioisosteric replacements
Anne Mai
Wassermann
and
Jürgen
Bajorath
*
Department of Life Science Informatics, B-IT, LIMES Program Unit Chemical Biology and Medicinal Chemistry, Rheinische Friedrich-Wilhelms-Universität, Dahlmannstr. 2, D-53113, Bonn, Germany. E-mail: bajorath@bit.uni-bonn.de; Fax: +49-228-2699-341; Tel: +49-228-2699-306
First published on 26th April 2011
Abstract
Bioisosteres are generally defined as similar chemical groups whose replacement in active compounds retains their biological activity. As such, bioisosteric replacements are of high interest in medicinal chemistry and bioisosterism continues to be an intensely investigated topic. Herein we have investigated the previously unexplored question as to whether bioisosteric replacements can be identified for individual target families. A total of 40 protein families were analyzed. Through a systematic compound data mining effort, we have identified 67 replacements of chemical groups that qualified as bioisosteres for only one target family. These bioisosteric replacements included groups of rather different sizes and chemical compositions and were directed against a total of 12 different target families including, among others, very popular targets such as tyrosine kinases or nuclear hormone receptors. A compendium of target family directed bioisosteric replacements is provided to aid in compound optimization efforts.
Introduction
In medicinal chemistry, bioisosteric replacements of functional groups in active compounds are highly desired to further increase the compound potency or improve other molecular properties.1,2 Bioisosteres are generally defined as pairs of groups with similar structure and properties whose replacement retains the biological activity of compounds. Hence, bioisosteric replacements represent conservative substitutions of chemical groups that are tolerated by biological targets but have the potential to modulate compound properties in a desired way. The search for bioisosteres has been ongoing since their original introduction in the 1950s3 and known or proposed bioisosteres have been compiled4 and organized in different databases.5,6 Given their practical utility for lead optimization projects, the interest in bioisosteres continues to be high.7In addition to chemical experience and intuition, computational methods have also been utilized to identify or predict bioisosteric replacements including both knowledge-based and ab initio approaches.7,8 In a recent study, we have applied the Matched Molecular Pair (MMP) concept9 to systematically search for potential bioisosteres in publicly available active compounds.10 MMPs are defined as pairs of compounds that only differ at a single site and are distinguished by a defined substituent or molecular fragment. Thus, we adapted the MMP concept as a generally applicable and consistent structural reference frame for the search for bioisosteric replacements in active compounds. In our initial study, we have identified a set of 96 general bioisosteric replacements for different compound activity classes.10 A subset of these replacements was previously not considered as potential bioisosteres.
Thus far, bioisosteric replacements have essentially been generalized, i.e. they have been considered to be bioisosteric across different targets, assuming that they might be conservative with respect to different biological activities. In compound optimization, one typically considers bioisosteres on the basis of this premise. A question that has not yet been investigated, but that is also of high relevance for medicinal chemistry applications, is whether or not bioisosteric replacements can be found that preferentially act against a given target family. Considering the individual structural constraints that must be met to yield specific target–ligand interactions, it would perhaps not be unlikely that such replacements might exist, although they are currently unknown.
In order to address this question we have carried out a large-scale data mining effort to search for replacements that act as bioisosteres in individual target families. For this purpose, we have applied the MMP formalism and implemented a search strategy specifically designed to identify target family directed bioisosteric replacements. The results of our analysis are reported herein.
Methods
Compound datasets
We extracted molecules with activity annotations against human target proteins from ChEMBL,11 a major public domain repository for bioactive compounds. Only potency measurements were selected with highest target confidence level (i.e. target confidence score 9 of the ChEMBL classification scheme) for direct target–ligand interactions (i.e. target relationship type “D”). Two types of potency measurements were considered. If Ki values were reported for a ligand–target combination, these were selected; otherwise, IC50 values were extracted, if available. Measurements containing threshold values (i.e., reported as “>” or “<”) were not considered because our approach to identify bioisosteres involved the calculation of potency differences between pairs of compounds (for which measurements with threshold values were not suitable). For compounds with multiple potency values reported against the same target, the arithmetic mean was calculated to yield a final potency. If individual measurements differed by more than one order of magnitude, all measurements were disregarded. From these molecules with fluctuating potency annotations, we calculated that 3202 different pair-wise assay combinations with significant measurement deviations existed.
Ligand sets were assembled for targets for which at least five active compounds were available meeting our criteria, leading to the selection of a total of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 868 compounds organized in 342 individual target sets. Furthermore, the 342 targets were grouped into 40 different target families following the sequence-based family annotation of Uniprot12,13 as well as the protein classification hierarchy of the ChEMBL database (for further dividing G-protein coupled receptors into families). All 40 target families investigated in this study are listed in Table 1. The number of targets per family ranged from 3 to 35. From all compounds, hierarchical scaffolds were extracted.14
868 compounds organized in 342 individual target sets. Furthermore, the 342 targets were grouped into 40 different target families following the sequence-based family annotation of Uniprot12,13 as well as the protein classification hierarchy of the ChEMBL database (for further dividing G-protein coupled receptors into families). All 40 target families investigated in this study are listed in Table 1. The number of targets per family ranged from 3 to 35. From all compounds, hierarchical scaffolds were extracted.14
| Target family | Source |
|---|---|
| a The 40 target families for which bioisosteric replacements were investigated are reported. Sources of target assignments to a given family are given under “Source”. | |
| AGC Ser/Thr kinase | UniProt |
| Aldo/keto reductase | UniProt |
| CAMK Ser/Thr kinase | UniProt |
| α-Carbonic anhydrase | UniProt |
| Chemokine receptor | ChEMBL |
| CMGC Ser/Thr kinase | UniProt |
| Cyclic nucleotide phosphodiesterase | UniProt |
| Cytochrome P450 | UniProt |
| Fatty-acid binding protein (FABP) | UniProt |
| Glutamate-gated ion channel (TC 1.A.10.1) | UniProt |
| Histone deacetylase | UniProt |
| Lipid-like ligand receptor | ChEMBL |
| Lipoxygenase | UniProt |
| Metabotropic glutamate receptor | ChEMBL |
| Monoamine receptor | ChEMBL |
| MPI phosphatase | UniProt |
| NOS | UniProt |
| Nuclear hormone receptor | UniProt |
| Nucleotide-like ligand receptor | ChEMBL |
| Peptidase A1 | UniProt |
| Peptidase C1 | UniProt |
| Peptidase C14A | UniProt |
| Peptidase M10A | UniProt |
| Peptidase M12B | UniProt |
| Peptidase S1 | UniProt |
| Peptidase S9B | UniProt |
| Phospholipase A2 | UniProt |
| PI3/PI4 kinase | UniProt |
| Potassium channel | UniProt |
| Protein-tyrosine phosphatase | UniProt |
| SDF symporter (TC 2.A.23) | UniProt |
| Short-chain dehydrogenase/reductase (SDR) | UniProt |
| Secretin-like receptor (B1) | UniProt |
| Ser/Thr kinase | UniProt |
| Short peptide receptor | ChEMBL |
| Sirtuin | UniProt |
| SNF symporter (TC 2.A.22) | UniProt |
| TKL Ser/Thr kinase | UniProt |
| Type-B carboxylesterase/lipase | UniProt |
| Tyrosine-protein kinase | UniProt |
Matched molecular pairs
For each target set, MMPs were identified using an in-house implementation15 of the Hussain and Rea algorithm.16 As illustrated in Fig. 1, an MMP is a pair of compounds that differ only at a single site, e.g. a specific R-group or ring system. Therefore, the compounds forming the MMP are related to each other by a defined substructure exchange or chemical “transformation”. For example, the compounds shown in Fig. 1 are inter-convertible by a “carboxylate to chlorine” transformation. However, the same MMP can be characterized by multiple transformations of different size. Hence, another valid transformation that describes the compound pair in Fig. 1 is a “p-benzoate to p-chlorophenyl” transformation. Hence, the applied algorithm might yield alternative, differently sized transformations for compound pairs. A total of 22631 of our 25868 source compounds formed one or more MMPs. Only 2.7% of all MMPs were based on the 3202 different pair-wise assay combinations with significant measurement deviations, as discussed above. These 2.7% of all MMPs were excluded from further analysis.
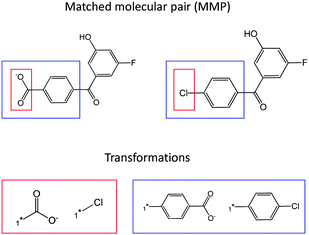 | ||
| Fig. 1 Matched molecular pair concept. An exemplary MMP is shown. The fragment that is exchanged between compounds forming a pair defines a transformation that relates the two MMP compounds to each other. The same MMP can be characterized by multiple transformations of different size. | ||
Target family directed bioisosteric replacements
The search protocol for bioisosteric replacements in target families consisted of the following steps:(1) For each transformation found in a target family compound set, all MMPs were assembled. MMPs corresponding to a transformation were required to be active against at least two targets of a family. The absolute logarithmic potency difference between compounds forming each MMP (“potency record”) was determined for all targets. Transformations were only considered if at least 20 potency records were available.
(2) MMPs representing a transformation were also required to contain at least five different scaffold pairs (hence, transformations had to occur in different structural contexts). In addition, if the target or the scaffold pair for which the largest number of potency records were available was removed from the analysis, at least 10 potency records had to remain.
(3) Potency differences within one order of magnitude were permitted for a transformation to qualify as a bioisosteric replacement. Only 1/15 of all available potency records were allowed to fall outside this potency range. Otherwise, the transformation was not further considered.
(4) If transformations of different size described the same MMPs, only the transformation describing the largest substructure was retained. However, if a smaller transformation was found in more MMPs than a larger one and if it also covered all compound pairs representing the larger transformation, then the smaller transformation was selected.
The assignment of transformations, MMPs, and potency records is illustrated in Fig. 2. All calculations reported herein were carried out with in-house generated Perl and Scientific Vector Language (SVL)17 programs and Pipeline Pilot18 tools.
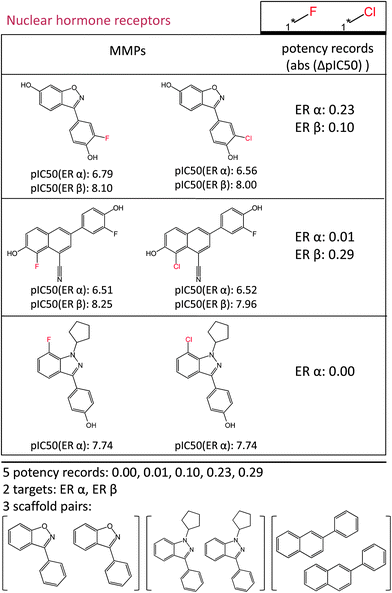 | ||
| Fig. 2 Assignment of MMPs and potency records to transformations. For each transformation occurring in the target family compound collection, all corresponding MMPs are assembled. The absolute logarithmic potency difference between compounds forming each MMP (“potency record”) is determined for all targets the MMP is active against. Here, three MMPs active against the nuclear hormone receptor family are defined by a “chlorine to fluorine” replacement and are grouped together. Their absolute logarithmic potency differences for the estrogen receptor alpha (ERα) and the estrogen receptor beta (ERβ) are also reported. In this example, five potency records for two different targets are obtained and the MMPs represent three different scaffold pairs. | ||
Results and discussion
Searching for target family directed bioisosteres
On the basis of our search strategy, transformations were regarded as potential bioisosteric replacements at the level of a target family if they were consistently represented by multiple MMPs with moderate potency differences for more than one target, if they occurred in different structural environments, and if the corresponding potency records were not significantly biased by a single target or scaffold pair.Potency variations as a consequence of transformations were largely (but not exclusively) limited to an order of magnitude. It should be noted that the analysis is cut-off sensitive. By considering smaller or larger potency variations, different numbers of target-family directed bioisosteres would be identified. However, in our opinion, a range of one order of magnitude is consistent with the basic idea of bioisosterism because a replacement resulting in a 100- or 1000-fold reduction in compound potency would hardly be regarded as bioisosteric. On the other hand, a ten-fold increase in potency as a consequence of a replacement would certainly be considered a favorable bioisosteric effect. Focusing our search on target families, we also required that a replacement had to occur in multiple ligands active against different family members and, in addition, that the potency record distribution was not strongly biased towards an individual target or chemotype.
Importantly, due to the general sparseness of currently available activity annotations, one would not be able to conclude with certainty that replacements meeting these selection criteria would be true bioisosteres for all targets belonging to a family, or that they could not act on a target belonging to another family. Therefore, replacements that ultimately met our criteria for only one target family were classified as target family directed (rather than family specific) bioisosteres.
Pre-selected transformations
For all compounds active against 40 target families, a total of 251638 (in part redundant) transformations were defined by MMPs, as illustrated in Fig. 1. This pool of transformations provided the starting point for our analysis. After applying our selection criteria, only 83 non-redundant transformations remained. The selection procedure traced transformations back to 16 target families. However, for a subset of transformations, multiple family assignments were obtained. We found more than one family assignment for a total of 16 transformations that are shown in Fig. 3. These transformations involved replacement of small functional groups (e.g.halogen atoms versusether groups), aliphatic linkers of different lengths, and phenyl rings with different substituents. Most of these transformations were assigned to two target families and, in a few instances, also to three or four. The extreme case has been a rather generic replacement of a meta- versus para-substituted phenyl ring, which was bioisosteric for ligands of six target families.
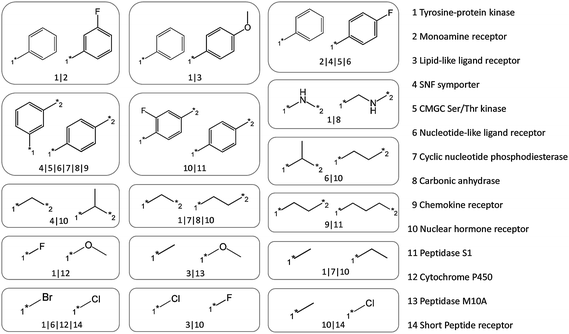 | ||
| Fig. 3 Bioisosteres directed at multiple target families. The 16 bioisosteric replacements that were identified for multiple target families are shown and annotated with their family assignments. | ||
Target family directed bioisosteric replacement
After removal of these 16 transformations, a total of 67 transformations remained that met our single target family constraint and hence qualified as target family directed bioisosteres. These 67 bioisosteric replacements were directed against 12 target families. Table 2 reports the distribution of bioisosteric replacements over these families. A total of 22 replacements were found in ligands active against the nucleotide-like ligand receptor family, which represented the largest number, followed by the short peptide receptor with 19 bioisosteric replacements, and the monoamine receptor, tyrosine kinase, and peptidase M10A families with five bioisosteric replacements, respectively. Table 2 also shows that there was no correlation between the number of targets per family and the number of detected replacements. While 5 bioisosteric replacements were found for inhibitors of tyrosine kinases, only a single bioisostere was detected for inhibitors of AGC serine/threonine kinases. In three more cases including the lipid-like ligand receptor, the SNF symporter (TC 2.A.22) and the peptidase C1 families, only a single qualifying replacement was identified. In Fig. 4a, all 67 target family directed bioisosteric replacements are shown. As can be seen, chemically different and differently sized replacements were observed for individual protein families.| TF | Abbr. | #Bioisosteres | #Targets |
|---|---|---|---|
| a Target families for which family directed bioisosteres were identified are listed in the column “TF” and are abbreviated (“Abbr.”). For each family the number of directed bioisosteres (“#Bioisosteres”) and the number of targets in the family (#Targets) are reported. | |||
| Nucleotide-like ligand receptor | NLR | 22 | 5 |
| Short peptide receptor | SPR | 19 | 35 |
| Peptidase M10A | M10A | 5 | 9 |
| Tyrosine-protein kinase | TK | 5 | 30 |
| Monoamine receptor | MAR | 5 | 34 |
| α-Carbonic anhydrase | CA | 3 | 9 |
| Peptidase S1 | S1 | 2 | 17 |
| Nuclear hormone receptor | NHR | 2 | 18 |
| SNF symporter (TC 2.A.22) | SNF | 1 | 5 |
| AGC Ser/Thr kinase | AGC | 1 | 14 |
| Peptidase C1 | C1 | 1 | 6 |
| Lipid-like ligand receptor | LLR | 1 | 20 |
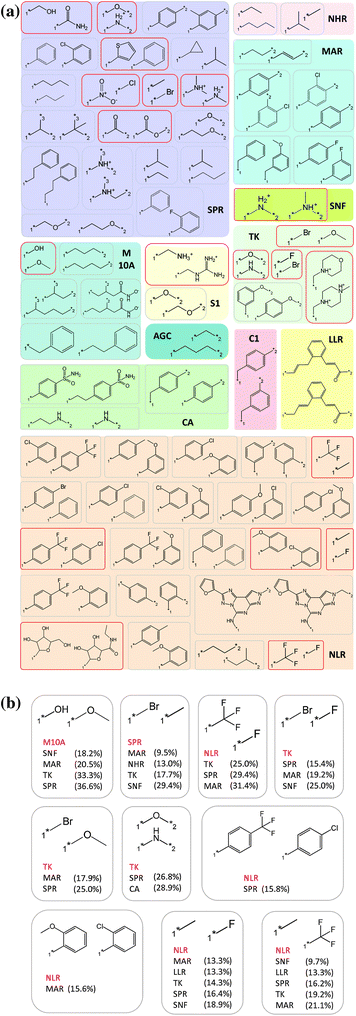 | ||
| Fig. 4 Target family directed bioisosteric replacements. (a) All 67 single target family directed bioisosteric replacements are shown and annotated with their family assignment. Bioisosteric replacements that involve exchanges of well-defined functional groups are marked in red. (b) Target family directed replacements are shown that would have qualified as bioisosteres for more than one target family if potency changes larger than one order of magnitude would have been accepted. The qualifying target family is abbreviated in red and other families that did not meet the potency criterion are shown and annotated with the relative frequency with which the transformation introduced potency changes larger than one order of magnitude. Abbreviations are used according to Table 2. SNF stands for the sodium neurotransmitter symporter family. | ||
Compound data mining efforts are generally affected by “data sparseness” (i.e., available compounds have not been tested on all families). Due to data sparseness, one cannot conclude with certainty that replacements identified for a single target family could not in principle act as bioisosteres on another target family. It is important to note that one can only extract information that currently available compound data provide. Therefore, we deliberately use the term “target family directed” (rather than “target family specific”) bioisosteres. However, in many instances, replacements that might, at first glance, look rather generic are indeed target-family directed because of the potency differences associated with them in different families. As an example, we consider the methyl to trifluoromethyl replacement that was found to be directed against the nucleotide-like ligand receptor family. This substitution was also frequently observed for monoamine receptors, tyrosineprotein kinases, short peptide receptors, lipid like ligand receptors, and SNF symporters. However, for these families, the replacement often induced large potency differences. For these families, potency changes of more than one order of magnitude were observed with frequencies of 21.1, 19.2, 16.2, 13.3, and 9.7%, respectively. Hence, in these cases, the methyl to trifluoromethyl substitution did not qualify as a bioisostere.
Chemical interpretation
A strength of the MMP-based approach to bioisostere analysis is that it requires no pre-conceived chemical notion of groups that might be of interest, but rather systematically detects all substructure exchanges that are defined by single transformations. Consequently, qualifying replacements were of rather different chemical nature involving not only functional groups but also linker fragments or substituted ring systems. Thus, for practical compound optimization, some target family directed bioisosteres involving well-defined functional groups might be of higher interest than others such as linker fragments, although these fragments also met all formal requirements for bioisosterism. In Fig. 4a, target family directed bioisosteres are highlighted that represent replacements of functional groups at specific positions, which would likely be preferred candidates for practical applications. In addition, Fig. 4b shows a subset of these preferred bioisosteres that would have also qualified for one to five other target families if potency changes of more than one order of magnitude would have been permitted in our analysis. However, their bioisosterism with respect to the secondary target families is in part questionable, due to large potency alterations.Conclusions
In this study, we have investigated a previously unexplored question concerning bioisosterism and systematically searched for bioisosteric replacements that were preferentially directed against individual target families. To these ends, we have employed the MMP concept to generalize our search and designed a specific computational bioisostere selection procedure for systematic compound data mining. Starting from a pool of 251638 candidate transformations extracted from MMPs active against 40 target families, we ultimately identified a set of 67 transformations each of which was directed against one of 12 protein families. Thus, target family directed bioisosteric replacements were indeed found in currently available active compounds. Target family directed bioisosteres were most frequently observed for the nucleotide-like ligand receptor and short peptide receptor families. The corresponding transformations were in part of rather different chemical nature. For drug discovery projects, the notion of such target family directed bioisosteric replacements should be helpful to support compound optimization efforts for different protein families, including pharmaceutical targets of high interest. Hence, for different targets, specific compound substitutions can be preferentially considered. The systematic compound data mining approach reported herein is generally applicable to search for bioisosteric replacements and other chemical transformations with desired properties and does not involve any proprietary components. The search can be readily extended and the search protocol can also be easily modified to adjust selection criteria for different applications. Taken together, the results presented herein suggest that the currently available repertoire of bioisosteric replacements might be further extended through systematic compound data analysis and that a subset of bioisosteres is preferentially directed against individual target families.
References
- P. H. Olesen, Curr. Opin. Drug Discovery Dev., 2001, 4, 471–478 Search PubMed.
- S. R. Langdon, P. Ertl and N. Brown, Mol. Inf., 2010, 29, 366–385 Search PubMed.
- H. L. Friedman, N. A. S.-N. R. C., Publ., 1951, 206, 295 Search PubMed.
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS.
- M. Devereux, P. L. Popelier and I. M. McLay, J. Chem. Inf. Model., 2009, 49, 1497–1513 Search PubMed.
- BIOSTER database. http://www.digitalchemistry.co.uk/, accessed September, 2010.
- P. Ertl, Curr. Opin. Drug Discovery Dev., 2007, 10, 281–288 Search PubMed.
- M. Devereux and P. L. Popelier, Curr. Top. Med. Chem., 2010, 10, 657–668 Search PubMed.
- P. W. Kenny and J. Sadowski, in Chemoinformatics in Drug Discovery, ed. T. I. Oprea, Wiley-VCH, Weinheim, Germany, 2005, pp. 271–285 Search PubMed.
- A. M. Wassermann, Future Med. Chem., 2011, 3, 425–436 Search PubMed.
- ChEMBL. http://www.ebi.ac.uk/chembl/, accessed May, 2010.
- UniProt. http://www.uniprot.org/, accessed September, 2010.
- UniProtConsortium, Nucleic Acids Res., 2010, 38, D142–D148 CrossRef.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS.
- A. M. Wassermann and J. Bajorath, J. Chem. Inf. Model., 2010, 50, 1248–1256 Search PubMed.
- J. Hussain and C. Rea, J. Chem. Inf. Model., 2010, 50, 339–348 Search PubMed.
- MOE (Molecular Operating Environment), Chemical Computing Group Inc., Montreal, Quebec, Canada, 2009.
- Scitegic Pipeline Pilot, Student Edition, Version 6.1, Accelrys, Inc., San Diego, CA, 2009.
| This journal is © The Royal Society of Chemistry 2011 |