DOI:
10.1039/C1MD00023C
(Concise Article)
Med. Chem. Commun., 2011,
2, 611-615
N1-Benzyl substituted cambinol analogues as isozyme selective inhibitors of the sirtuin family of protein deacetylases†
Received
21st January 2011
, Accepted 21st March 2011
First published on 5th May 2011
Abstract
The human deacetylase SIRT2 is believed to promote neurodegeneration with recent studies demonstrating that a reduction in the activity of SIRT2 can rescue alpha synuclein toxicity in Parkinson's disease models. In contrast, a second member of the sirtuin family, SIRT1, is believed to play a neuroprotective role. This dichotomy places an additional challenge in the path of sirtuin inhibitor development as a need for isozyme selectivity arises. By combining computational methods with assessment of the biological activity of novel N1-substituted cambinol analogues, further insights that are relevant to this challenge are obtained.
The type III NAD+-dependent protein deacetylases, known as the sirtuins, remain a target of considerable interest for a range of therapeutic applications.1 Whilst studies initially focused on the discovery of sirtuin activators, reports on the development of novel inhibitors have also appeared.2 The high degree of interest in the sirtuins stems from their ability to deacetylate both histone and non-histone proteins including p53.3 p53 is a known substrate of SIRT1 and the activity of p53 has been proposed to be downregulated by SIRT2 in an AKT dependent manner.4 Of the seven human enzymes, it is the development of inhibitors of SIRT1 and SIRT2 that has received the most attention.2 Key roles for SIRT2 in cell cycle regulation, due to its ability to deacetylate tubulin, and in the control of nervous system function have recently been proposed.5 SIRT2 is believed to promote neurodegeneration and a selective inhibitor of SIRT2, AGK2 (1) (Fig. 1), has been shown to rescue alpha synuclein toxicity in a Drosophila model of Parkinson's disease.6a AGK2 (1) has also been shown to provide neuroprotection in models of Huntington's disease through inhibition of SIRT2.6b Interestingly, SIRT1 is thought to play a neuroprotective role,5b raising further interest in highly potent and selective SIRT2 inhibitors.
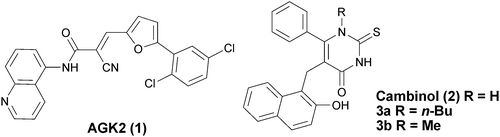 |
| | Fig. 1 Structures of AGK-2 (1), cambinol (2) and N1-n-butyl 3a and N1-methyl 3b substituted cambinol analogues. | |
We have recently reported that it is possible to tune the essentially unselective sirtuin inhibitor, cambinol (2) generating a more potent and selective SIRT2 inhibitor through incorporation of an alkyl substituent at the N1-position.7 For example, N1-n-butyl substituted cambinol 3a was 6-fold more active against SIRT2 than SIRT1 in vitro. In addition, experiments in cells analysing p53 induction and tubulin acetylation supported the view that 3a inhibits SIRT2 but not SIRT1.7a Based on preliminary molecular modelling studies, we proposed that the increase in potency of 3a against SIRT2 may result from differences in the ability of the two enzymes to incorporate the N1-alkyl side chain of 3a into the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnicotinamide exchange tunnel leading to increased stabilisation of the SIRT2-3a complex compared to the SIRT1-3a complex.7a Whilst this tunnel has been described,8 the possibility that it could be used to influence inhibitor selectivity has not been exploited. As it continues to prove challenging to access nanomolar non-peptidic inhibitors of SIRT2, we decided to design and prepare additional N1-substituted cambinol analogues. The results of this study are presented, starting with a higher level molecular modelling study. In the SIRT1-3a complex, residue Gln296 undergoes a change during the course of the simulation, relative to the conformation at the start of the simulation, and stabilises a COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond between Gln296 and the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydroxyl group of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring in 3a. An analogous event is seen during the simulation of the SIRT2-3a complex, this time involving Glu116 (see movies in the ESI†).9b Any disruption of these COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bonds will likely disrupt the binding of an inhibitor to either protein.11 Furthermore the central COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundthiouracil ring in 3a appears to interact with the neighbouring Gln167 and His187 residues in the SIRT2-3a complex. Gln167 (OE1) appears to form a COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond with the N3H of 3a whilst His187 may form a COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond with the C4-carbonyl COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundoxygen.
Further analysis of the sirtuin-compound 3a complexes
Our previously reported SIRT2 selective inhibitor 3a was analysed using extensive molecular docking and molecular dynamics methods. Evolution of the secondary structures of the SIRT2-3a model during the simulations showed that the amino acid residues around the C-pocket (Ile93-Gly102, referred to here as Loop 1) that connect helices α2 and α3 exist as a loop in the SIRT2-3a model (Fig. 2a) whilst the same region in the SIRT1-3a model assumes a helical conformation (Fig. 2b and associated movies in the ESI†).9b This arises from the differences in the sequence in Loop 1 between SIRT2 (93IPDFRSPSTG102) and SIRT1 (IPDFRSRDG), notably the absence of a Pro (P99) in SIRT1. It is well established that COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundproline can act as a helix breaker.10 The change in the conformation of this region of the protein from a loop (in SIRT2) to a helix (in SIRT1) makes the active site in SIRT1 more open and shallow compared to that in SIRT2.7a This also results in Arg97 in SIRT2 covering the active site with the hydrophobic part of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundArg sidechain. In SIRT1, the analogous COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundarginine (Arg276, not shown) is located on the helix and is exposed to solvent. In the SIRT2-3a complex, π–π interactions lead to the packing of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring in 3a against H187 and F119, providing a compact fit (Fig. 2A) whilst in the SIRT1-3a complex the “more open” pocket provides a larger space for 3a to move around, resulting in a less-packed structure (Fig. 2B). In fact the equivalent His residue (His365) in SIRT1 flips its sidechain during the simulations and the final location of 3a is different to that in the SIRT2-3a complex.9b
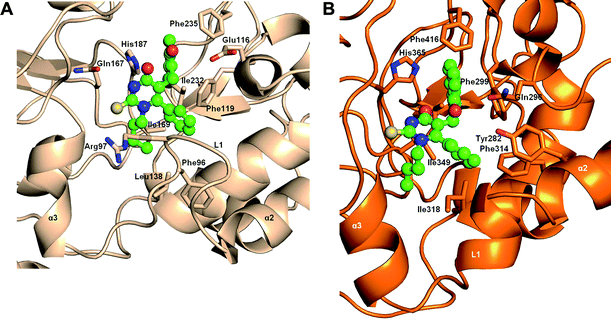 |
| | Fig. 2 Models of cambinol analogue 3a bound to A) human SIRT2 and B) human SIRT1. Key residues involved in the binding of the ligand are shown in sticks. These snapshots were taken at the end of 10 ns of molecular dynamics simulation (carried out using the programme Amber9 as described in ESI page S2†) and show that the loop region in SIRT2 is stable while the loop region in SIRT1 is helical leading to a more open binding pocket in SIRT1. Docking of cambinol analogues was carried out prior to simulations as described in ESI page S2.† | |
Binding free energy analysis9a
The energetics associated with the changes over the time course of the two simulations were computed (Tables S1 and S2, ESI†).9b As expected from the structural differences observed during the simulations, a more negative ΔGbind (∼10 kcal mol−1) characterises the association between SIRT2 and 3a. A breakdown of the energetic contributions shows that the major differences arise from the van der Waals and electrostatic energies, with the former dominating. Indeed, if the penalty for burying charges is taken into account, the electrostatic contributions are destabilising. This is understandable given that 3a buries several charges that are located in the active site (the polar atoms of active site residues and Glu116/Gln296). Tables S3 and S4 (ESI†) list the residues that contribute most to the binding energetics and, as expected, the major contributions arise from the active site residues (there are only 6 residues contributing >kT in SIRT1 whilst there are 8 in SIRT2; Fig. 2A and B). In both systems the maximum contributions (>2.5 kT) were from a Phe (299 and 119 in SIRT1 and SIRT2 respectively) and an COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundIle (349 and 169 in SIRT1 and SIRT2 respectively).
Novel N1-benzyl substituted cambinol analogues
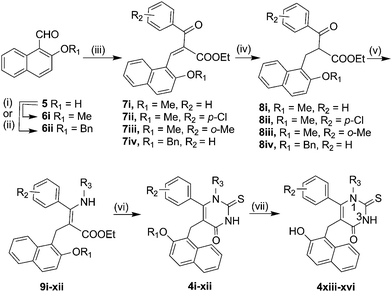
As the higher level computational studies supported our previously proposed binding mode for 3a,7a it was decided to assess whether alternative N1-substituted cambinol analogues may have improved interactions in this region. A preliminary visual inspection suggested that changes to the N1-substitutent in 3a may be accommodated. It was proposed that a N1-benzyl substituent might be acceptable in place of the N1-n-butyl in 3a. In addition, a para-substituent on the N1-benzyl group was proposed to be positioned such that it could enter into the exchange tunnel, potentially stabilising the complex further. As the incorporation of a range of p-alkoxy substituents on the benzyl ring was viewed as synthetically achievable, our attention focused on analogues of the general type 4 (Scheme 1 and Table 1) including 4xv (Fig. 3).
 |
| | Scheme 1 Synthesis of cambinol analogues 4i–xvi. See Table 1 for substituent structures. Reagents and conditions: (i) MeI, K2CO3, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMF, rt, 18 h; (ii) BnBr, K2CO3, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundacetone, reflux, 18 h; (iii) ethyl benzoyl acetates, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundpiperidine, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundEtOH, 18 h, reflux; (iv) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundNaBH4, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundpyridine, rt, 2 h; (v) R3NH2, CH3COOH, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundethanol, 2 d, reflux (due to instability, these enamines were reacted directly in the following cyclisation reaction); (vi) TMSNCS, 3 h, reflux; (vii) BCl3, n-Bu4NI, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDCM, −78 to 0 °C, 2 h. | |
Table 1 Inhibition at 60 μM ± SEa of SIRT1 and SIRT2 by the novel analogues 4i–xvi, 10 and 11i–v.
| Compound |
R1 |
R2 |
R3 |
SIRT1e |
SIRT1e IC50/μM |
SIRT2 |
SIRT2e IC50/μMb |
|
SE, standard error (n = 2).
IC50 values against the two enzymes were determined for compounds with greater than 70% inhibition at 60 μM against SIRT2.
Controls.
See Scheme 2.
For details of final enzyme concentrations see ESI.†
n.d. not determined.
|
|
2
|
H |
H |
H |
59.5 ± 1 |
40.7 ± 117a |
51.9 ± 1 |
47.9 ± 127a |
|
3b
|
H |
H |
Me |
29.4 ± 1 |
> 907a |
73.7 ± 1 |
20 ± 57a |
|
4i
|
Me |
H |
Me |
36.2 ± 5 |
— |
37.7 ± 2 |
— |
|
4ii
|
Me |
p-Cl |
n-Bu |
18.4 ± 7 |
— |
49.4 ± 5 |
— |
|
4iii
|
Me |
o-Me |
n-Bu |
11.5 ± 1 |
— |
37.3 ± 2 |
— |
|
4iv
|
Me |
H |
Bn |
n.d.f |
— |
37.7 ± 11 |
— |
|
4v
|
Me |
H |
p-MeOBn |
8.8 ± 1 |
— |
66.1 ± 4 |
— |
|
4vi
|
Me |
H |
p-EtOBn |
19.3 ± 1 |
> 120 |
72.2 ± 2 |
16 ± 4 |
|
4vii
|
Me |
H |
p-ProBn |
38.0 ± 1 |
61.9 ± 2 |
71.8 ± 1 |
11 ± 2 |
|
4viii
|
Me |
H |
p-BuOBn |
32.7 ± 2 |
— |
66.1 ± 1 |
— |
|
4ix
|
Bn |
H |
Bn |
25.4 ± 2 |
— |
48.4 ± 3 |
— |
|
4x
|
Bn |
H |
p-MeOBn |
29.8 ± 2 |
— |
56.9 ± 1 |
— |
|
4xi
|
Bn |
H |
p-EtOBn |
36.6 ± 1 |
— |
67.2 ± 4 |
— |
|
4xii
|
Bn |
H |
p-ProBn |
22.8 ± 3 |
— |
65.1 ± 5 |
— |
|
4xiii
|
— |
H |
Bn |
10.8 ± 4 |
— |
62.0 ± 1 |
— |
|
4xiv
|
— |
H |
p-MeOBn |
n.d. |
58.2 ± 4 |
73.4 ± 2 |
12 ± 1 |
|
4xv
|
— |
H |
p-EtOBn |
13.4 ± 8 |
> 120 |
88.9 ± 1 |
4 ± 1 |
|
4xvi
|
— |
H |
p-ProBn |
15.6 ± 6 |
> 120 |
85.4 ± 2 |
6 ± 3 |
|
10
|
H |
— |
Bn |
1.8 ± 2 |
— |
27.0 ± 9 |
— |
|
11i
|
Bnd |
— |
Bn |
23.0 ± 4 |
— |
45.1 ± 1 |
— |
|
11ii
|
Bnd |
— |
p-MeOBn |
32.2 ± 4 |
— |
67.0 ± 2 |
— |
|
11iii
|
Bnd |
— |
p-EtOBn |
35.0 ± 4 |
68.1 ± 5 |
73.3 ± 1 |
10 ± 6 |
|
11iv
|
Bnd |
— |
p-BuOBn |
8.5 ± 1 |
— |
63.3 ± 1 |
— |
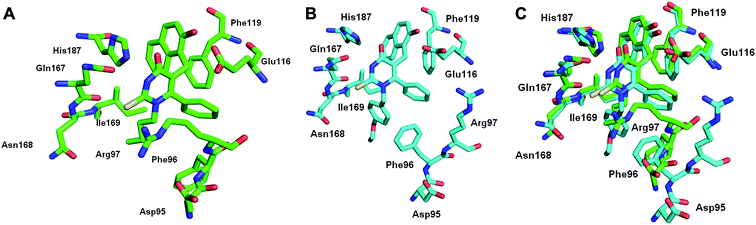 |
| | Fig. 3 Close-up view of the proposed binding mode of A) 3a and B) 4xv in SIRT2; C) an overlap of the two calculated binding modes. | |
Using analogous methods to those used for modelling the SIRT2-3a complex, an analysis of the binding mode of 4xv to SIRT2 was carried out (Fig. 3). Encouragingly, the ΔGbind was, within error, the same for the two complexes and the two N1-substituents were predicted to occupy similar positions, although the p-alkoxy substituent in 4xv appears to reach further into the exchange tunnel in the SIRT2-4xv complex. A key difference between the two predicted complexes is the position of Arg97. In the SIRT2-3a complex, the hydrophobic component of the side chain packs against the N1-n-butyl substituent forming a computationally stable interaction. In the case of the simulation of the SIRT2-4xv complex, Arg97 undergoes a series of conformational changes alternating between packing against the ligand and moving out into solvent. This is reflected in the lower average interaction energy for Arg97 in the SIRT2-4xv complex (Table S8, ESI†). Given the challenges that still exist in the synthesis of highly potent and selective SIRT2 inhibitors and the questions raised by the computational studies, it was decided to test whether incorporation of a N1-p-substituted benzyl substituent in 2 would lead to a further stabilisation of the ligand-SIRT2 complexes compared to 3a.
Synthesis of novel N1-substituted cambinol analogues
The scope of our reported route to N1-substituted analogues proved to be limited to linear alkyl groups.7a We therefore developed a modified protocol to prepare analogues 4i-xvi (Scheme 1). The COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol functionality in 5 was protected as either a methyl or a COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundbenzyl ether giving naphthaldehydes 6i and 6ii respectively. Treatment of 6i and 6ii with different ketoesters afforded intermediates 7i–iv, which underwent selective reduction with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundNaBH4 in COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundpyridine to give 8i–iv. Reaction of 8i–iv with a range of different amines in the presence of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundacetic acid afforded enamines of general structures 9i–xii, which underwent cyclisation to give the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundthiouracil ring on treatment with neat TMSNCS (4i–xii).12 The structure of 4iv was confirmed by small molecule X-ray crystallographic studies. In the case of the O-benzyl protected analogues, deprotection occurred on treatment with BCl3 and nBu4NI at low temperatures to give 4xiii–xvi.13 A similar approach was used to prepare analogues in which the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring was replaced by (i) a COMPOUND LINKS
Read more about this on ChemSpider
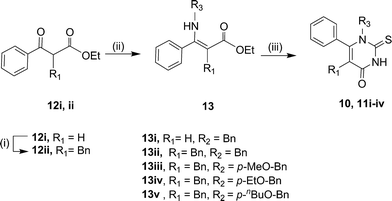
Download mol file of compoundhydrogen (10); (ii) a benzyl substituent (11i–iv, Scheme 2). Benzylation of the α-position of 12i afforded compound 12ii.14 Subsequent treatment of 12i with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundbenzylamine and 12ii with different amines under acidic conditions afforded enamines 13i–v. Cyclisation with neat TMSNCS gave analogues 10 and 11i–iv.
In vitro inhibition of SIRT1 and SIRT2 by analogues 4i–xvi, 10 and 11i–v
All the cambinol analogues were tested for in vitro activity against SIRT1 and SIRT2 using an established fluorescent assay system (Table 1).7a Replacement of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring present in control compound 3b (Scheme 1) with a methoxy-substituted COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring (to give 4i, Scheme 1 and Table 1) resulted in a significant loss of inhibitory activity against SIRT2, with 4i showing only 38 ± 2% inhibition of SIRT2 at a final concentration of 60 μM (cf.3b, 74 ± 1% under identical conditions). Similarly, 4ii and 4iii, two analogues that contain an N1-n-butyl substituent as well as additional substituents in the C6-aromatic ring, and the N1-benzyl substituted analogues 4iv and 4ix showed poor inhibitory activity against both enzymes. However, the situation was rescued to some extent by the incorporation of a para-alkoxy substituent in the N1-benzyl group with reasonably active analogues being prepared despite the fact that they contained either a methoxy- (4v–4viii) or a benzyloxy- (4x–4xii) substituent in the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring. For example, 4vi, an analogue that contains a para-n-propoxyl substituent in the N1-benzyl ring and a methoxy substituent in the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring, had an IC50 = 11 ± 2 μM against SIRT2. A further increase in the p-alkoxy chain length led to a small decrease in the activity against SIRT2 (4viii). All the analogues 4i–xii were poor inhibitors of SIRT1. The low activity against SIRT2 of 4i–xii is consistent with the predicted formation of a complex-stabilising COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond between the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol OH group of 3a and Glu116 in SIRT2. The absence of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundOH group in 4i–xii means that the inhibitors cannot act as COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond donors to Glu116, although they do still have the capacity to act as a COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond acceptors. When the ability to act as COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen bond donors to Glu116 was returned to this compound series (in 4xiii–4xvi) the most active analogues against SIRT2 were prepared. Interestingly, the incorporation of a para-alkoxy substituent in the N1-benzyl substituent again lead to an improvement in activity against SIRT2 with the most potent compound 4xv having an IC50 of 4 ± 1 μM. 4xv is equipotent with our previously reported SIRT2-selective inhibitor 3a and AGK-2 (1). The need for the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring was investigated further in a second series of analogues (10 and 11i–11iv). In these analogues the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol group was replaced with either a COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen (10) or a benzyl-substituent (11i–11iv). As expected, activity was lost in the absence of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring with 10 being inactive against both enzymes. Replacement of the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnaphthol ring with a C5-benzyl substituent (11i–11iv) again lead to a reduction in potency although an analogous trend in activity against SIRT2 was observed as a function of the N1-substituent with a p-ethoxybenzyl group again being optimal (11iii). Consistent with the in vitro results, 4xv led to a larger increase in acetylated tubulin levels in H1299 cells in the presence of Trichostatin A than cambinol (2) (fold increase in observed levels of acetylated tubulin in the presence of 60 μM 4xv or 2 was 1.7 and 1.3 respectively compared to the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO control). This assay was also used to show that 4xv and 3a increased acetylated tubulin levels by almost equivalent amounts (see Fig. S1 in the ESI†).
The search continues for inhibitors of SIRT2. Identification of potent and selective inhibitors of this isozyme remains important due to the need for chemical tools to study SIRT2 biology and the proposed links between SIRT2 inhibition and neurodegenerative disease. The incorporation of a liphophilic alkyl substituent at the N1-position in cambinol (2) was shown previously by us to lead to a significant increase in potency against SIRT2.7a Here, detailed molecular dynamics simulations supported our initially proposed inhibitor binding mode and showed a clear difference in stability between the SIRT1-3a and the SIRT2-3a complexes. These computational studies also suggested that inclusion of an N1-p-alkoxy-substituted benzyl substituent may lead to an increase in inhibitor potency by retaining interactions between the enzyme and the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundthiouracil core but enabling the N1-substituent to reach further into the COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundnicotinamide exchange tunnel. It was hoped that these factors would overcome potential problems envisaged due to predicted changes in the interaction of Arg97 with the ligand and that an overall stabilisation of the SIRT2-inhibitor complex would occur. In fact, after further development of our synthetic strategies to cambinol analogues, our studies showed that incorporation of this alternative N1-substituent lead to compounds (4xv and 4xvi) that were equipotent with our previously reported analogue 3a. One possible rationalisation is that any binding energy gained by 4xv as a result of reaching further into the tunnel is counteracted by a loss of interaction with Arg97. Our current work focuses on methods of obtaining the best of both worlds. Whilst the inclusion of a longer N1-alkyl substituent (e.g. hexyl) is tempting, it seems likely that the expected increase in entropic cost on binding associated with incorporation of this flexible chain and issues relating to cell permeability may prove problematic.
Notes and references
- For a recent review see J. Schemies, U. Uciechowska, W. Sippl and M. Jung, Med. Res. Rev., 2009, 30(6), 861–889 Search PubMed.
-
(a) F. J. Alcain and J. M. Villalba, Expert Opin. Ther. Pat., 2009, 19, 283–294 Search PubMed;
(b) Y. Cen, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1635–1644 CrossRef CAS.
- H. Lin, Org. Biomol. Chem., 2007, 5, 2541–2554 RSC.
- H. Jin, Y. Kim, D. Kim, K. Baek, B. Y. Kang, C. Yeo and K. Lee, Biochem. Biophys. Res. Commun., 2008, 368(3), 690–695 Search PubMed.
-
(a) B. J. North, B. L. Marshall, M. T. Borra, J. M. Denu and E. Verdin, Mol. Cell, 2003, 11, 437–444 CrossRef CAS;
(b) for a recent review see K. Harting and B. Knoll, Eur. J. Cell Biol., 2010, 89, 262–269 Search PubMed.
-
(a) T. F. Outeiro, E. Kontopoulos, S. M. Altmann, I. Kufareva, K. E. Strathearn, A. M. Amore, C. B. Volk, M. M. Maxwell, J. C. Rochet, P. J. McLean, A. B. Young, R. Abagyan, M. B. Feany, B. T. Hyman and A. G. Kazantsev, Science, 2007, 317, 516–519 CrossRef CAS;
(b)
R. Luthi-Carter; D. M. Taylor; J. Pallos; E. Lambert; A. Amore; A. Parker; H. Moffitt; D. L. Smith; H. Runne; O. Gokce; A. Kuhn; Z. M. Xiang; M. M. Maxwell; S. A. Reeves; G. P. Bates; C. Neri; L. M. Thompson; J. L. Marsh; A. G Kazantsev. Proc. Nati. Ac. Sci. USA, 107, pp. 7927–7932 Search PubMed.
-
(a) F. Medda, R. J. M. Russell, M. Higgins, A. R. McCarthy, J. Campbell, A. M. Z. Slawin, D. P. Lane, S. Lain and N. J. Westwood, J. Med. Chem., 2009, 52, 2673–2682 Search PubMed and references therein;
(b) B. Heltweg, T. Gatbonton, A. D. Schuler, J. Posakony, H. Li, S. Goehle, R. Kollipara, R. A. DePinho, Y. Gu, J. A. Simon and A. Bedalov, Cancer Res., 2006, 66, 4368–4377 Search PubMed.
-
(a) J. L. Avalos, J. D. Boeke and C. Wolberger, Mol. Cell, 2004, 13, 639–648 CrossRef CAS;
(b) PDB Id 2HJH.
-
(a) Docking studies alone are known to suffer from lack of conformational sampling and their force fields are fairly simplistic. We therefore carried out molecular dynamics simulations under explicit solvent conditions. The free energies computed using the MMGBSA methods are limited in their accuracies and yet have been demonstrated the be very useful for comparison of energetics;
(b) See ESI† for further information and the following URL for movies http://web.bii.a-star.edu.sg/bmad/cambinol-2011/.
- A. Chakrabartty, T. Kortemme and R. L. Baldwin, Protein Sci., 1994, 3(5), 843–852 CAS.
- A. J. Tervo, S. Kyrylenko, P. Niskanen, A. Salminen, J. Leppanen, T. H. Nyronen, T. Jarvinen and A. Poso, J. Med. Chem., 2004, 47, 6292–6298 CrossRef CAS.
- F. D. Therkelsen, A. L. L. Hansen, E. B. Pedersen and C. Nielsen, Org. Biomol. Chem., 2003, 1, 2908–2918 RSC.
- P. R. Brooks, M. C. Wirtz, M.
G. Vitelino, D. M. Rescek, G. F. Woodworth, B. P. Morgan and J. W. Coe, J. Org. Chem., 1999, 64, 9719–9721 CrossRef CAS.
- S. Jacobson and B. Grosh, J. Chem. Soc. Trans., 1915, 107, 959–966 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic characterisation of all new compounds, movies of the molecular dynamics simulations, computational parameters and tubulin acetylation assay data have been provided. CCDC reference number 809750. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1md00023c |
| ‡ Authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.