Development of a phosphatase-resistant, L-tyrosine derived LPA1/LPA3 dual antagonist
James E. East†
*a,
Karen M. Carter†a,
Perry C. Kennedyb,
Nancy A. Schultec,
Myron L. Toewsc,
Kevin R. Lynchb and
Timothy L. Macdonalda
aDepartment of Chemistry, University of Virginia, PO Box 400319, McCormick Road, Charlottesville, VA 22904, USA. E-mail: je5y@virginia.edu; Tel: +1-434-924-0595
bDepartment of Pharmacology, University of Virginia School of Medicine, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA
cDepartment of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, 985800 Nebraska Medical Center, Omaha, NE 68198-5800, USA
First published on 3rd March 2011
Abstract
Lysophosphatidic acid (LPA) is a bioactive compound that has gained attention due to its role in neoplastic diseases. Our group has developed a potent dual LPA1/LPA3 receptor antagonist, VPC51098 (LPA1 IC50 = 84 nM, LPA1 IC50 = 48 nM) that contained a labile phosphate head group. This lability has impaired our evaluation of our scaffold of LPA receptor antagonistsin vivo. We wished to replace the phosphate with a potentially more stable head group while retaining potency at both LPA1 and LPA3 to facilitate future in vivo studies. We tested in vitro potency of all head groups including α-methylene, α-fluoromethylene, α-hydroxymethylene; vinyl phosphonates; α-fluoro vinyl phosphonates. The most potent compound was found to be a low micromolar inhibitor VPC51299 that contained a vinyl phosphonate and possessed a half-life of approximately 90 min in rats when dosed intravenously. Herein, we describe the synthesis and initial biological evaluation of these compounds.
Lysophosphatidic acid (LPA) is a glycerophospholipid found in all eukaryotic cells as an intermediary metabolite, but it is also found in mammalian plasma at about 500 nM. Extracellular LPA was recognized initially for its role in the regulation of blood pressure.1 There are now numerous established physiological roles for extracellular LPA, including angiogenesis,2 motility, proliferation3 and survival4 of diverse cell types. Because of its role in angiogenesis and cell growth, LPA has garnered attention as a potential therapeutic target in neoplastic disease.5,6,7,8 Further, studies with mice lacking functional LPA1 receptor alleles have implicated LPA in the development of pulmonary fibrosis9 and neuropathic pain.10
LPA is synthesized from lysophosphatidylcholine (LPC) by the plasma lysophospholipase D enzyme autotaxin (ATX).11,12LPA interacts with a set of at least five G protein-coupled receptors (GPCRs), LPA1-5. These receptors are responsible for the mitogenic and migratory properties assigned to LPA.13 Detailed descriptions of LPA functions and the roles of specific receptors have been reviewed elsewhere.14
Our laboratories have developed LPA analogs with a particular focus on antagonists of the LPA receptors. Our earliest series of LPA analogs featured the N-acyl ethanolamide phosphoric acid (NAEPA) backbone, which was a modification of the natural glycerol moiety in LPA.15 The NAEPA compounds eventually evolved into LPA analogs containing various amino acids16 with D-tyrosine analogs exhibiting the best activities at the LPA receptors.17Phosphate head group mimetics, including phosphonic and thiophosphonic acid derivatives,18 were also investigated, because of their enhanced metabolic stability.
The most promising of this series of compounds, VPC51098, contained a 4-(2,2,2-trifluoroethoxy)pyridine moiety (Fig. 1). In vitro assays showed the compound to have nanomolar affinity, yet whole animal studies were hampered due to the instability of the phosphatein vivo, which presumably was due to hydrolysis catalyzed by the ubiquitous lipid phosphate phosphatases (LPPs).19 In this paper we report the synthesis of a hydrolytically stable vinyl phosphonate derivative that is an antagonist at LPA1 and LPA3 receptors. This compound should prove useful as a tool to further study the biological functions of LPA1 and LPA3in vivo.
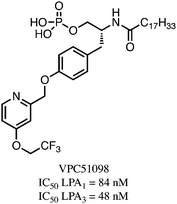 | ||
| Fig. 1 LPA1/LPA3 dual antagonist VPC51098. | ||
Replacing the bridging oxygen of the phosphate with a carbon atom changes the electronic character and, as a result, the acidity of the head group. Studies investigating the second deprotonation of phosphates and phosphonates have demonstrated a pKa of 6.2 for the former and 7.6 for the latter.20 This difference in electronic make-up and acidity can affect enzyme-substrate interactions. For example, phosphonate analogs that inhibit glycerol 3-phosphate dehydrogenase have been shown to bind in their dianionic state, whereas their monoanionic states act neither as substrates nor as inhibitors.21 To best mimic the phosphate head group of LPA, we considered it approximate the pKa2 of the phosphate. This can be achieved by attaching electron-withdrawing groups α and/or β to the phosphorus atom. With this in mind, we synthesized a series of saturated and unsaturated phosphonates bearing electron-withdrawing groups.
The synthesis of the vinyl phosphonate began with the sequential Boc and silyl protection of D-tyrosine methyl ester that allowed for the reduction to the tyrosinal by DIBAL-H to give 2 (Scheme 1). Horner–Wadsworth–Emmons olefination with either tetraethyl methylenediphosphonate or tetraethyl (fluoromethylene)bis(phosphonate) yielded protected vinyl phosphonates 3.1 and 3.2. TBAF deprotection of the silyl ether set up a Williamson ether synthesis with (4-(2,2,2-trifluoroethoxy)pyridin-2-yl)methyl methanesulfonate 20 to yield 4.1 and 4.2. TFA mediated Boc deprotection followed by treatment with palmitoyl chloride gave amides 5.1 and 5.2. Deprotected products 6.1 and 6.2 were realized through the treatment of precursor compounds with TMSBr and MeOH hydrolysis of silyl ethers. Protected vinyl phosphonate 5.1 was reduced using hydrogenolysis and then deprotected with TMSBr to afford phosphonate 8 (Scheme 2).
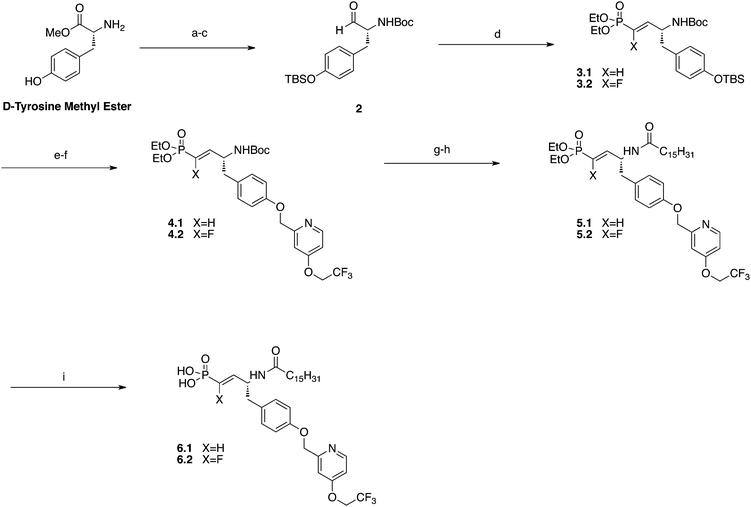 | ||
| Scheme 1 Preparation of vinyl phosphonates. Reagents and conditions: (a) Boc2O, DIEA, DCM, rt. (b)TBSCl, Imid., DCM, rt. (c) DIBAL-H, DCM, −78 °C. (d) Tetraethylmethylene phosphonate or fluorotetramethylene phosphonate, NaH, THF, 0 °C, 43%. (e) TBAF, DCM, 0 °C to rt. (f) K2CO3, 18-crown-6, 20, acetone, reflux, 36–51%. (g) TFA, DCM, rt. (h) palmitoyl chloride, DIEA, DCM, 0 °C, 51–55%. (i) TMSBr, DCM, 0 °C to rt, 92–93%. | ||
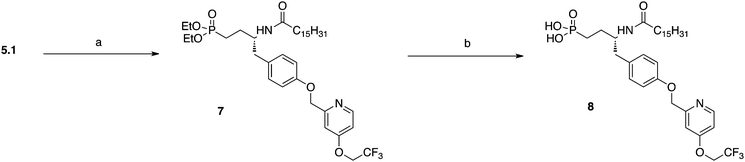 | ||
| Scheme 2 Preparation of phosphonate. Reagents and conditions: (a) H2, Pd/C, EtOH, rt., 91%. (b) TMSBr, DCM 0 °C to rt, 38%. | ||
Fluoro-vinyl phosphonate 9 was achieved after deprotection of the Boc group and the formation of the amide with palmitoyl chloride (Scheme 3). TBAF deprotection followed by hydrogenation of the double bond yielded α-fluoromethylene phosphonates 10a and 10b. The individual diastereomers were separated by flash chromatography prior to deprotection but the stereochemistry for each compound was not determined. The pyridyl ethers of each diastereomer were achieved through Williamson ether synthesis to give 11a and 11b. Finally TMSBr deprotection gave α-fluoromethylene phosphonates 12a and 12b.
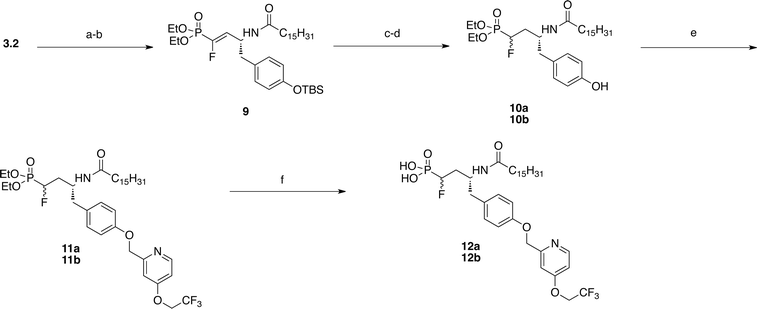 | ||
| Scheme 3 Preparation of α-fluoro phosphonates. Reagents and conditions: (a) TFA, DCM, rt. (b) palmitoyl chloride, DIEA, DCM, 0 °C to rt., 38%. (c) TBAF, DCM, 0 °C to rt. (d) H2, Pd/C, MeOH, 29%. (e) 20, K2CO3, 18-crown-6, acetone, reflux, 73–75%. (f) TMSBr, DCM, 0 °C to rt, 74–83%. | ||
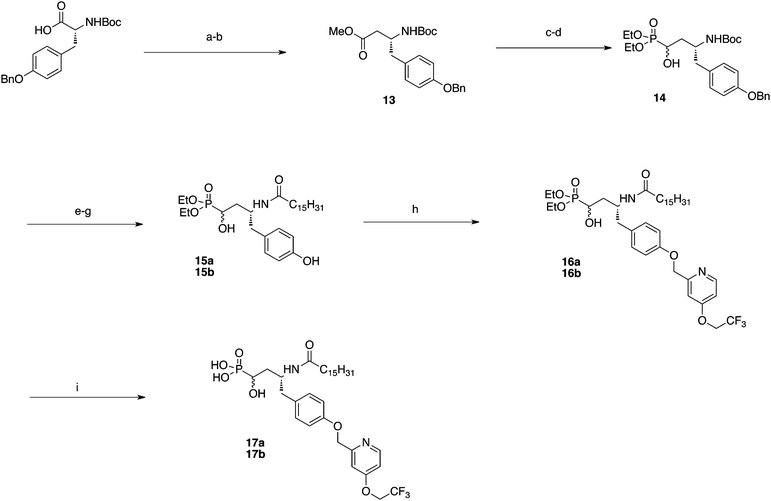
The α-hydroxy phosphonates were also synthesized to assess their electron-withdrawing ability. N-Boc, O-Benzyl protected D-tyrosine was subjected to diazomethane homologation (Scheme 4) to give methyl ester 13. The ester was treated with DIBAL-H and the resulting aldehyde was reacted with diethylphosphite to give α-hydroxy phosphonate 14 as a mixture of diastereomers. Boc deprotection followed by amide formation and hydrogenolysis gave 15a and 15b. The pyridine group was attached through Williamson ether synthesis to give 16a and 16b. TMSBr deprotection afforded 17a and 17b.
 | ||
| Scheme 4 Preparation of α-hydroxy phosphonates. “17a” refers to first diastereomer to elute from column, “17b” refers to the later elute. Reagents and conditions: (a) isobutylchloroformate, THF, 0 °C, 97%. (b) CH2N2, silver benzoate, TEA, −78 °C, 93%. (c) DIBAL-H, DCM, 78 °C, 81%. (d) Diethylphosphite, NaH, THF, −78 °C, 31%. (e) TFA, DCM, 0 °C. (f) Palmitoyl chloride, DIEA, DCM, 0 °C to rt. (g) H2, Pd/C, rt, 85% overall. (h) 20, K2CO3, 18-crown-6, acetone, reflux, 89–91%. (i) TMSBr, DCM, 0 °C to rt, 14–28%. | ||
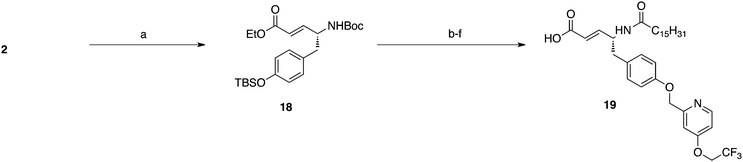
A vinyl carboxylic acid head group was also explored for its ability to mimic the natural phosphate pKa2. Horner–Wadsworth–Emmons olefination was performed on starting tyrosinal, 2, to give protected vinyl carboxylic 18 (Scheme 5). Following attachment of the acyl tail and the pyridyl moiety deprotection of the head group yielded the carboxylic acid, 19.
 | ||
| Scheme 5 Preparation of vinyl carboxylic acids. Reagents and conditions: (a) triethyl phosphonoacetate, NaH, THF, 0 °C, 50%. (b) TBAF, DCM, 0 °C, 72%. (c) K2CO3, 18-crown-6, acetone, 20, 72%. (d) TFA, DCM, 0 °C. (e) palmitoyl chloride, DIEA, DCM, 0 °C. (f) LiOH, EtOH:THF, rt, 40% overall. | ||
The compounds were initially screened on A431 human epithelial carcinoma cells for LPA-induced intracellular calcium flux (Fig. 2). These cells have been shown by real time polymerase chain reaction (RT/PCR) to express LPA1-3 receptors mRNAs and respond to LPA stimulation.8 All compounds were collided initially at a single concentration (10 μM) against a single concentration of 1-oleoyl LPA (30 μM). This initial screen showed that the vinyl phosphonate, 6.1; α-fluoro vinyl phosphonate, 6.2; and α-fluoro phosphonate, 12b were the most effective inhibitors of flux in A431 cells. Although it was initially thought that predicted acidity of the head group would correlate to activity there was no evidence of a trend of the pKa2 of the phosphate isostere with activity.22
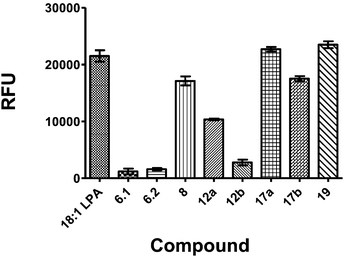 | ||
| Fig. 2 A431 calcium mobilization assay for initial determination of LPA receptor inhibition. A431 cells, expressing LPA receptors 1–3, were used to collide drug (10 uM) with LPA (10 uM). Inhibition of calcium release allowed for an initial determination of LPA receptor antagonism. | ||
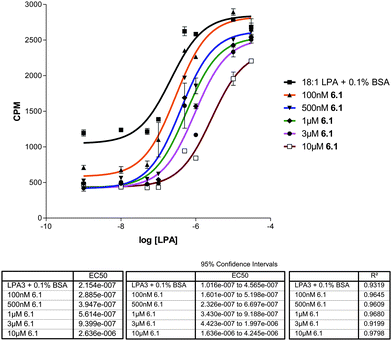
The most potent compound from the initial screen, 6.1, was assayed for LPA1 and LPA3 antagonism in a GTP[γ-35S] binding assay using membrane fractions from RH7777 cells over-expressing LPA1 (Fig. 3) and CHO cells over-expressing LPA3 (Fig. 4) to confirm inhibition of LPA receptor activity indirectly observed in Fig. 2. Increased concentrations of 6.1 resulted in a parallel rightward shift in the LPA concentration effect curve at LPA1 (Fig. 3) and LPA3 (Fig. 4), which indicates that 6.1 behaves as a competitive antagonist at these receptors. Schild analyses gave Ki values of 143 nM and 512 nM at LPA1 and LPA3, respectively. In contrast, 6.1 was not active at LPA2 (data not shown). The potency of 6.1 was indistinguishable from that of the benchmark LPA1/3 antagonist, Ki16425 (not shown).8
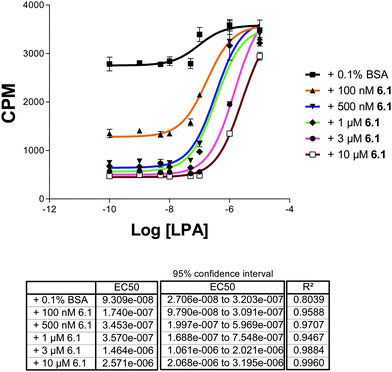 | ||
| Fig. 3 Dose-dependent inhibition of hLPA1 receptor activity by 6.1. Membrane fractions containing hLPA1 from CHO cells were used to create a dose response curve with 6.1 over varying concentrations of drug. Schild analysis was derived from this data. | ||
 | ||
| Fig. 4 Dose-dependent inhibition of hLPA3 receptor activity by 6.1. Membrane fractions containing hLPA3 from CHO cells were used to create a dose response curve with 6.1 over varying concentrations of drug. Schild analysis was derived from this data. | ||
Vinyl phosphonate 6.1 was validated as an LPA1/LPA3 antagonist by evaluating its ability to block the LPA-driven inhibition of β agonist-stimulated cAMP accumulation in rat glioma C62B cells. Isoproterenol is a β adrenoreceptor agonist that increases cAMP levels in many cells. LPA, acting via one or more of its Gi-coupled receptors, which include LPA1 and LPA3, almost completely inhibited the isoproterenol stimulation of cAMP in C62B cells. Addition of an LPA receptor antagonist together with isoproterenol and LPA is thus expected to restore cAMP accumulation to the isoproterenol-stimulated values observed in the absence of LPA. Phosphonate 6.1 indeed behaved in this manner, i.e. antagonism of this LPA receptor response in a dose-dependent fashion was observed (Fig. 5). As observed with recombinant receptors in broken cell assays, addition of increasing concentrations of antagonist resulted in parallel, rightward shifts of the dose response curves (Fig. 6). Schild analysis of these data yielded a Ki value of approximately 11 nM, which is in the range of values determined with LPA1 and LPA3 receptors. Finally, a known agonist of the LPA3 receptor, 21 (Fig. 7),23 was co-administered with 6.1 to determine if 6.1 could block agonism at LPA3. By measuring calcium release it was found that 6.1 did indeed overcome agonism mediated by compound 21 in a dose-dependent fashion (Fig. 8). In addition 6.1 showed a half-life of 90 min in rats when dosed intravenously (see the ESI)
![cAMP accumulation in C62B cells upon treatment with isoproterenol, 18 : 1 LPA, and 6.1. C62B cells were grown to confluence and then incubated for 1 h with [3H]adenine to label cellular ATP pools. Cells were then stimulated for 2 min with isoproterenol (10 μM) in the absence or presence of LPA (100 nM) or related compounds to inhibit cAMP accumulation, each in the absence or presence of 6.1 or other antagonists to block the LPA inhibition.](/image/article/2011/MD/c0md00273a/c0md00273a-f5.gif) | ||
Fig. 5 cAMP accumulation in C62B cells upon treatment with isoproterenol, 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 LPA, and 6.1. C62B cells were grown to confluence and then incubated for 1 h with [3H]adenine to label cellular ATP pools. Cells were then stimulated for 2 min with isoproterenol (10 μM) in the absence or presence of LPA (100 nM) or related compounds to inhibit cAMP accumulation, each in the absence or presence of 6.1 or other antagonists to block the LPA inhibition. :1 LPA, and 6.1. C62B cells were grown to confluence and then incubated for 1 h with [3H]adenine to label cellular ATP pools. Cells were then stimulated for 2 min with isoproterenol (10 μM) in the absence or presence of LPA (100 nM) or related compounds to inhibit cAMP accumulation, each in the absence or presence of 6.1 or other antagonists to block the LPA inhibition. | ||
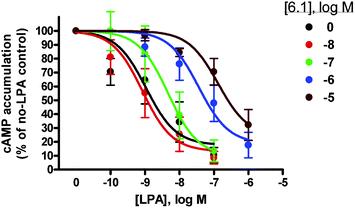 | ||
| Fig. 6 Dose response curve of 6.1 in C62B cells. | ||
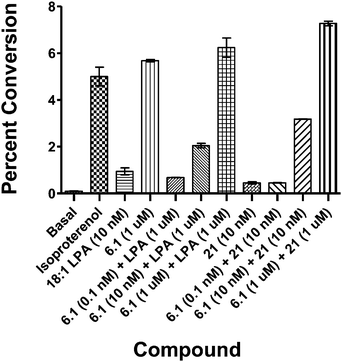 | ||
| Fig. 8 Reversal of 21-generated agonism by 6.1 using the isoproterenol cAMP accumulation assay. In a similar fashion to Fig. 5, C62B cells were stimulated with isoproterenol in the absence or presence of LPA (100 nM) or related compounds to inhibit cAMP accumulation, each in the absence or presence of 6.1 or agonist 21 to block or stimulate the LPA inhibition. | ||
Small molecule antagonists of the LPA receptors continues to attract attention by virtue of the putative role of LPA in pathologies such as neoplastic diseases, tissue fibrosis and neuropathic pain. For these applications, potent and long-lived compounds are desirable, for their ability to be used in long term in vivo studies and for the hoped-for eventual use in human medicine. Herein, we describe the synthesis and in vitro characterization of several phosphatase-resistant analogs of our antagonist, VPC51098.
All of the analogs were initially screened in an A431 calcium mobilization assay. Compounds 6.1, 6.2 and 12b proved to be the most potent in this initial assay. 6.1 was found to be a potent inhibitor of LPA1 and LPA3, with Ki values of 143 nM and 512 nM respectively. 6.1 was taken as a lead compound into a C62B cell-based assay to further assess receptor antagonism. Not only were the previous assay results verified, but also it was found that 6.1 was able to overcome the agonism displayed by known LPA1 agonist 21.
With the discovery of the potent LPA1/LPA3 dual antagonist 6.1 it will now be possible to probe the long-term effects of blocking the LPA1 and LPA3 signaling pathways in animal models of various human diseases. In addition to moving this compound into animal models, another important goal will be to develop antagonist analogs selective for only LPA1 and only LPA3.
References
- S. Sen, R. Smeby and F. M. Bumpus, Am. J. Physiol., 1968, 214, 337 Search PubMed.
- C. Rivera-Lopez, A. Tucker and K. Lynch, Angiogenesis, 2008, 11, 301 CrossRef CAS.
- E. J. van Corven, A. Groenink, K. Jalink, T. Eichholtz and W. H. Moolenaar, Cell, 1989, 59, 45 CrossRef CAS.
- J. S. Levine, J. S. Koh, V. Triaca and W. Lieberthal, Am. J. Physiol., 1997, 273, F575 CAS.
- Y. Xu, G. Jiang, R. Tsukahara, Y. Fujiwara, G. Tigyi and G. Prestwich, J. Med. Chem., 2006, 49, 5309 CrossRef CAS.
- G. Durgam, T. Virag, M. Walker, R. Tsukahara, S. Yasuda, K. Liliom, L. van Meeteren, W. Moolenaar, N. Wilke, W. Siess, G. Tigyi and D. Miller, Synthesis, J. Med. Chem., 2005, 48, 4919 CrossRef CAS.
- U. Hasegawa, J. Erickson, G. Goddard, S. Yu, S. Liu, K. Cheng, A. Eder, K. Bandoh, J. Aoki, R. Jarosz, A. Schrier, K. Lynch, G. Mills and X. Fang, J. Biol. Chem., 2003, 278, 11962 CrossRef CAS.
- H. Ohta, K. Sato, N. Murata, A. Damirin, E. Malchinkhuu, J. Kon, T. Kimura, M. Tobo, Y. Yamazaki, T. Watanabe, M. Yagi, M. Sato, R. Suzuki, H. Murooka, T. Sakai, T. Nishitoba, D. Im, H. Nochi, K. Tamoto, H. Tomura and F. Okajima, Mol. Pharmacol., 2003, 64, 994 CrossRef CAS.
- A. Tager, P. LaCamera, B. Shea, G. Campanella, M. Selman, Z. Zhao, V. Polosukhin, J. Wain, B. Karimi-Shah, N. Kim, W. Hart, A. Pardo, T. Blackwell, Y. Xu, J. Chun and A. Luster, Nat. Med., 2008, 14, 45 CrossRef CAS.
- J. Nagai, H. Uchida, Y. Matsushita, R. Yano, M. Ueda, M. Niwa, J. Aoki and J. Chun, Mol. Pain, 2010, 6, 78 CrossRef.
- A. Tokumura, E. Majima, Y. Kariya, K. Tominaga, K. Kogure, K. Yasuda and K. Fukuzawa, J. Biol. Chem., 2002, 277, 39436 CrossRef CAS.
- M. Umezu-Goto, A. Kishi, K. Taira, N. Hama, K. Dohmae, T. Takio, G. Yamori, K. Mills, J. Inoue and H. Aoki, J. Cell Biol., 2002, 158, 227 CrossRef CAS.
- J. Chun, T. L. Hla, K. R. Lynch, W. Moolenaar and S. Spiegel, Pharmacol. Rev., 2010, 62, 579 CrossRef CAS.
- J. Choi, D. Herr, K. Noguchi, Y. Yung, C. Lee, T. Mutoh, M. Lin, S. Teo, K. Park, A. Mosley and J. Chun, Ann. Rev. Toxicology, 2010, 50, 157 Search PubMed.
- S. Hook, S. Ragan, D. Hopper, C. Honemann, M. Durieux, T. Macdonald and K. Lynch, Mol. Pharmacol., 1998, 53, 188.
- B. Heasley, R. Jarosz, K. Lynch and T. Macdonald, Bioorg. Med. Chem. Lett., 2004, 14, 2735 CrossRef CAS.
- B. Heasley, R. Jarosz, K. Carter, S. Van, K. Lynch and T. Macdonald, Bioorg. Med. Chem. Lett., 2004, 14, 4069 CrossRef CAS.
- W. Santos, B. Heasley, R. Jarosz, K. Carter, K. Lynch and T. Macdonald, Bioorg. Med. Chem. Lett., 2004, 14, 3473 CrossRef CAS.
- S. Hooks, W. Santos, D. Im, H. Heise, T. Macdonald and K. Lynch, J. Biol. Chem., 2001, 276, 4611 CrossRef CAS.
- J. Nieschalk, A. Batsanov, D. O'Hagan and J. Howard, Tetrahedron, 1992, 52, 165.
- P. Adams, R. Harrison and T. Inch, Biochem J., 1974, 141, 729 CAS.
- C. Rye and J. Baell, Curr. Med. Chem., 2005, 12, 3127 CrossRef CAS.
- M. Panchatcharam, S. Miriyala, F. Yang, M. Rojas, C. End, C. Vallant, A. Dong, K. Lynch, J. Chun, A. Morris and S. Smyth, Circ. Res., 2008, 103, 662 CrossRef CAS.
Footnote |
| † These authors contributed equally to this manuscript |
| This journal is © The Royal Society of Chemistry 2011 |