DOI:
10.1039/C0MD00261E
(Concise Article)
Med. Chem. Commun., 2011,
2, 966-972
Design, synthesis and biological evaluation of γ-lactam hydroxamate based TACE inhibitors†‡§
Received
15th December 2010
, Accepted 6th May 2011
First published on 13th June 2011
Abstract
A new series of γ-lactam hydroxamate based TACE inhibitors was designed mainly by introducing various substitutions at the 2nd position of the quinoline nucleus to achieve high potency and good selectivity towards TACE over COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundmatrix metalloproteases (MMPs) and ADAM-10. In ex vivo TNF-α inhibitory activity assays, compounds 11o and 11p were identified as the most potent compounds. The in vitro TACE inhibitory activity, selectivity over MMPs and ADAM-10 and the in vivo TNF-α inhibitory activities of compounds 11o and 11p were assessed and lead compound 11p was identified. Preliminary toxicity and pharmacokinetic (PK) studies were conducted for compound 11p and it showed an improved PK and clean toxicological profile compared to standard compound 1. Altogether, these results demonstrated the discovery of highly potent and selective γ-lactam hydroxamate based TACE inhibitors which show potential for the safe and effective treatment of inflammatory diseases.
Introduction
Tumor Necrosis Factor-α (TNF-α) is a potent proinflammatory cytokine and it plays a central role in host defence and inflammation.1 Human TNF-α is a 26 kDa membrane bound precursor (tmTNF) that is cleaved by a cell membrane associated zinc-metalloprotease ADAM17 (A Disintegrin And Metalloprotease), also known as TNF-α converting enzyme (TACE) to form a 17 kDa soluble TNF-α (sTNF-α). In general, TNF binds to two receptor subtypes (TNFR1 and R2) preferentially, TNFR1 is activated by sTNF-α and TNFR2 by tmTNF.2,3 An excessive production of sTNF-α is a recognized pathogenic mediator in a number of acute and chronic inflammatory conditions including rheumatoid arthritis (RA) and Crohn's disease.4–6
Inhibition of TACE activity to control the level of sTNF-α offers a promising therapeutic approach for the treatment of inflammatory diseases and several orally active small molecule based TACE inhibitors are reported in the literature.7–10 While designing these TACE inhibitors, attempts were made to achieve good selectivity against closely associated matrix metalloproteases (MMPs).11,12 Despite the fact that two TACE inhibitors (BMS-561392 (1) and TMI-005 (2); Fig. 1) were advanced to Phase II clinical trial stages, so far no TACE inhibitor reached the market, mainly because of hepatotoxicity and/or lack of efficacy.13,14 The exact reasons for the development of hepatotoxicity and lack of efficacy in humans are still not clear, but it must be emphasized that the earlier TACE inhibitors discovery efforts were mainly directed to achieve selectivity over MMPs and limited attention has been paid to discriminate between TACE and other ADAM proteases.13–15 Among various ADAM family members, ADAM-10 has the highest amino acid sequence homology with TACE (especially in catalytic domain) and it also triggers catalytic conversion of tmTNF to sTNF-α.16–19
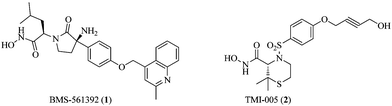 |
| | Fig. 1 Structures of TACE inhibitors under clinical developments (BMS-561392 and TMI-005). | |
The docking studies and X-ray crystal structure of TACE co-crystallized with γ-lactam hydroxamate based TACE inhibitors revealed that the aromatic moiety (COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundquinoline nucleus at P1 region) occupies the S1 site of the enzyme, the oxygen atom of the pyrrolidinone ring forms hydrogen bonds with L348 and G349 and the isobutyl group occupies the small hydrophobic pocket (S2). The hydroxamic acid interacts with the zinc atom present in the active site of TACE and coordinates with the side-chain carboxylate of E406, while the phenyl ring exhibits aromatic stacking with the imidazole side chain of H405 (Fig. 2).20,21
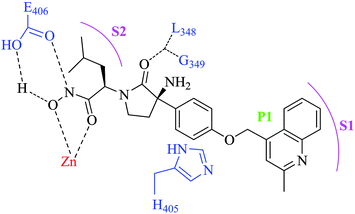 |
| | Fig. 2 Pictorial representation of key interactions of BMS-561392 (1) with corresponding binding sites of TACE. | |
As the S1 site of TACE is larger and bend-shaped, with respect to most of the MMPs and ADAM-10, substitution with bulky groups at P1 region of TACE inhibitors (such as 2-methylquinolin-4-yl-methoxy group, in BMS-561392) is known to result in higher potency as well as good selectivity towards TACE over MMPs.21–23 The SAR studies of sulfonamide based TACE inhibitor scaffolds demonstrated that selectivity for TACE over MMPs and ADAM-10 were greatly enhanced due to an alkynyl group at P1 position (such as 4-hydroxybutynyl group in TMI-005), which specifically was accommodated into the S1 site of TACE, mainly due to the acquisition of its favorable bent confirmation.19,24–26 Thus, the difference in the shape and size of the S1 pocket of TACE over MMPs and ADAM-10 could be exploited to design potent and selective TACE inhibitors devoid of any MMPs and ADAM-10 activity.27 As a part of our ongoing research on TACE inhibitors28 and taking above structural features into consideration, in the present communication a new series of γ-lactam hydroxamate based TACE inhibitors were designed mainly by introducing various substituents at the 2nd position of the quinoline nucleus to achieve high potency and good selectivity towards TACE over MMPs and ADAM-10.
Results and discussion
Chemistry
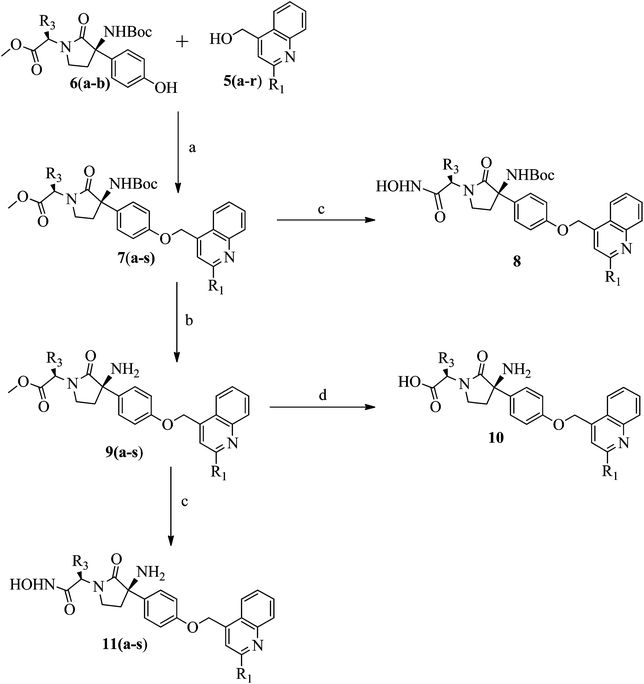
Synthesis of titled compounds (8–10 and 11a–s) was carried out as depicted in Scheme 1 and 2. The starting materials, 2-substiuted quinoline-4-carboxylic acid derivatives (3a–r) were synthesized as per the literature procedure.28,29 These acids were converted to corresponding methyl esters (4a–r) by treating with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundthionyl chloride (SOCl2) in COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compounddichloromethane (DCM), Scheme 1. The methyl esters of compounds 4a–r were reduced into corresponding 2-substituted quinolin-4-yl-methanol (5a–r), in the presence of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundsodium borohydride (NaBH4). Further compounds 5a–r were coupled with phenol derivatives (6a–b),30 under Mitsunobu conditions (DEAD and TPP) to get the key intermediates (7a–s), Scheme 2. The ester group of compound 7o was converted into the corresponding hydroxamic acid derivative (8), using hydroxylammoniumchloride (NH2OH·HCl). The TFA mediated Boc-group deprotection of compounds 7a–s led to the formation of compounds 9a–s, with free amino groups. The hydrolysis of compound 9o with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundNaOH yields compound 10, with a free acid group, while treatment of compound 9a–s, with NH2OH·HCl gives corresponding hydroxamic acid derivatives (11a–s).
 |
| | Scheme 1 Reagents and conditions: a) SOCl2, CHCl2, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundMeOH; b) NaBH4, CH2Cl2. Wherein, 3, 4 & 5a–r: R1 = Ph–; 3Me–Ph–; 4Me–Ph–; 4Cl–Ph–; 4F–Ph–; 4MeO–Ph–; OBzl–; Cl–; CF3–; Et–; iPr–; Cpr–; Chex–; MeO–; MeO–Me–; MeO–Et–; iPrO–Me– and iPrO–Et– | |
Following Scheme 1 and 2, a total of 22 compounds (8–10 and 11a–s) were prepared in good yield, under the mild reaction conditions and the overall percentage yield were found to be in the range of 60–70%. The ESI MS showed molecular ion peaks [M+], at different intensities, corresponding with the molecular weights of titled compounds. The elemental analyses of all the titled compounds were found within the limit of ±0.04% of theoretical values. The spectral data of all the synthesized compounds were found to be in conformity with the structure assigned and the corresponding IR, 1H NMR and ESI-MS data are presented in experimental section (see ESI§).
Ex
vivo TNF-α inhibitory activity and structure activity relationship (SAR)
Ex
vivo TNF-α inhibitory activity (using human whole blood assay) was assessed, mainly to establish the structure activity relationship (SAR) of a new series of γ-lactam hydroxamate based TACE inhibitors (8, 9o, 10 and 11a–s). As depicted in Table 1, depending upon the nature of substitutions, all the titled compounds showed a different degree of TNF-α inhibition (IC50). Substitution with bulky and aromatic groups at the 2nd position of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundquinoline ring in compound 1, such as phenyl- (11a), 3-methyl phenyl- (11b) and OBzl- (11g) analogs showed weak ex vivo TNF-α inhibition. Test compounds substituted with electron withdrawing groups at para-position of aromatic ring system, such as 4-chloro phenyl (11d), 4-fluro phenyl (11e) and 4-methoxy phenyl (11f) showed moderate TNF-α inhibition, while substitution with electron donating groups, such as 4-methyl phenyl (11c) showed weak TNF-α inhibition. Similarly, direct substitution of electron withdrawing groups such as Cl (11h) and CF3 (11i) at the 2nd position of COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundquinoline ring showed moderate TNF-α inhibition, while substitution with electron donating groups, such as ethyl (11j), isopropyl (11k), cyclopropyl (11l) and cyclohexyl (11m) showed good TNF-α inhibition, whereas, substitution with a strong electron donating group, such as MeO (11n) showed moderate TNF-α inhibition.
Table 1
Ex
vivo TNF-α inhibition (IC50) activity of test compounds (8, 9o, 10 and 11a–s)a
| Comp. No |
R1 |
R2 |
R3 |
R4 |
IC50 (nM)b |
|
Ex
vivo TNF-α inhibition was carried out in human whole blood and plasma TNF-α concentration was determined using an ELISA kit.
IC50 values (nM) are from single determination and compound 1 (BMS-561392) was used as positive std. control.
|
|
8
|
MeO–Me– |
–NHBoc |
iBu– |
–NHOH |
3050 |
|
9o
|
MeO–Me– |
–NH2 |
iBu– |
–COOMe |
3000 |
|
10
|
MeO–Me– |
–NH2 |
iBu– |
–COOH |
3230 |
|
11a
|
Ph– |
–NH2 |
iBu– |
–NHOH |
3057 |
|
11b
|
3Me–Ph– |
–NH2 |
iBu– |
–NHOH |
3876 |
|
11c
|
4Me–Ph– |
–NH2 |
iBu– |
–NHOH |
2899 |
|
11d
|
4Cl–Ph– |
–NH2 |
iBu– |
–NHOH |
286 |
|
11e
|
4F–Ph– |
–NH2 |
iBu– |
–NHOH |
290 |
|
11f
|
4MeO–Ph– |
–NH2 |
iBu– |
–NHOH |
250 |
|
11g
|
OBzl– |
–NH2 |
iBu– |
–NHOH |
4000 |
|
11h
|
Cl– |
–NH2 |
iBu– |
–NHOH |
450 |
|
11i
|
CF3– |
–NH2 |
iBu– |
–NHOH |
470 |
|
11j
|
Et– |
–NH2 |
iBu– |
–NHOH |
111 |
|
11k
|
iPr– |
–NH2 |
iBu– |
–NHOH |
140 |
|
11l
|
Cpr– |
–NH2 |
iBu– |
–NHOH |
170 |
|
11m
|
Chex– |
–NH2 |
iBu– |
–NHOH |
177 |
|
11n
|
MeO– |
–NH2 |
iBu– |
–NHOH |
401 |
|
11o
|
MeO–Me– |
–NH2 |
iBu– |
–NHOH |
11 |
|
11p
|
MeO–Et– |
–NH2 |
iBu– |
–NHOH |
13 |
|
11q
|
iPrO–Me– |
–NH2 |
iBu– |
–NHOH |
123 |
|
11r
|
iPrO–Et– |
–NH2 |
iBu– |
–NHOH |
130 |
|
11s
|
MeO–Me– |
–NH2 |
Me– |
–NHOH |
98 |
|
1
|
|
|
|
|
60 |
Introduction of methoxy-methyl (11o) and methoxy-ethyl (11p) groups at the 2nd position of the quinoline ring resulted in extremely potent TNF-α inhibition compared to the standard compound 1 (IC50: 60 nM), with IC50 values of 11 and 23 nM respectively, while isopropoxy-methyl (11q) and isopropoxy-ethyl (11r) showed relatively less potency than methyl analogs (1). Interestingly, substitution of R3- (isobutyl group) with methyl group (11s) showed less potency than 11o, while replacement of R2- (amino group) with NHBoc and substitution of R4 (hydroxamic acid group) with either methyl-ester (9o) or carboxylic acid (10) resulted in complete loss of TNF-α inhibition activity, indicating that a free amino group at R2 position and hydroxamic acid at R4 position is mandatory for TNF-α inhibition.
In general, compounds with electron donating groups showed good TNF-α inhibition, electron withdrawing substitutions showed moderate TNF-α inhibition, while bulky and aromatic substitutions showed weak TNF-α inhibition. Although the S1 binding pocket of TACE is large, the substitution with aromatic and bulky groups was found to be unfavorable. On the contrary, substitution with methoxy-methyl (11o) and methoxy-ethyl (11p) groups at the 2nd position showed excellent TNF-α inhibition while isopropoxy-methyl (11q) and isopropoxy-ethyl (11r) showed relatively less potency than methyl analogs (1), which could be due to the favorable bent confirmation acquisition at S1 binding pocket.23 Overall, ex vivo TNF-α inhibition results reveal that the potency of γ-lactam hydroxamate based TACE inhibitors can be modulated using suitable substituents at the 2nd position of the quinoline ring system.
In vitro
TACE inhibitory activity and selectivity
The in vitro TACE inhibitory activity and selectivity over MMPs (MMP-1, -2, -3, -7, -8, -9, -13 and 14) and ADAM-10 were evaluated for the most potent compounds (11o and 11p), using a fluorescence-based FRET assay and IC50 values were determined (Table 2).24 Both the test compounds inhibits the recombinant TACE with IC50’s of 2.1 and 2.3 nM respectively, compared to standard compound 1 (IC50: 12 nM). Compounds 11o and 11p showed >500-fold selectivity (IC50: >1000 nM) over all tested MMPs and ADAM-10, whereas standard compound 1 showed variable selectivity (IC50: < 500 nM) over tested MMPs and it showed only 40-fold selectivity over ADAM-10. Together, these in vitro results demonstrated the improved potency and selective profile of our test compounds (11o and 11p) over standard compound 1, which could be attributed mainly due to suitable substituents at the 2nd position of the quinoline ring system.
Table 2
In vitro
TACE inhibitory activity and selectivity over MMPs and ADAM-10 for selected test compounds (11o–p and 1)a
| Enzyme# |
Compounds |
| Compd 1 (std) |
11o
|
11p
|
|
In vitro
TACE inhibitory activity and selectivity over MMPs (MMP-1, -2, -3, -7, -8, -9, -13 and 14) and ADAM-10 were evaluated for selected test compounds—11o, 11p and compound 1 (BMS-561392)—using fluorescence-based FRET assay and IC50 values (nM) were determined (n = 3).
|
|
TACE
|
12 |
2.1 |
2.3 |
|
ADAM-10
|
480 |
1211 |
1199 |
|
MMP-1
|
429 |
1132 |
1059 |
|
MMP-2
|
482 |
1167 |
1088 |
|
MMP-3
|
285 |
1082 |
1020 |
|
MMP-7
|
466 |
1242 |
1008 |
|
MMP-8
|
299 |
1010 |
1002 |
|
MMP-9
|
389 |
1156 |
1210 |
|
MMP-13
|
461 |
1230 |
1114 |
|
MMP-14
|
499 |
1067 |
1085 |
Docking studies
The molecular docking studies for 11p and BMS-561392 (1) were carried out on the TACE (pdb id: 2FV5)31 crystal structure using Glide (version 50207)32 of Schrodinger. The crystal structure 2FV5 was obtained from RCSB protein data bank33 and the molecules 11p and 1 were geometrically optimized by using the Ligprep module of Schrodinger. The aromatic moiety (COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundquinoline) of 11p occupies the S1 site of the enzyme pocket, the oxygen atom of the pyrrolidinone ring forms hydrogen bonds with L348 and G349, the hydroxamic acid interacts with the zinc atom present in the active site of TACE and coordinates with the side-chain carboxylate of E406. All the five binding poses obtained for 11p were found to be close, with an RMSD of <0.8 Å and there were no unusual conformations. The ligand orientation and H-bonding interactions of 11p matches closely with 1 (Fig. 3).
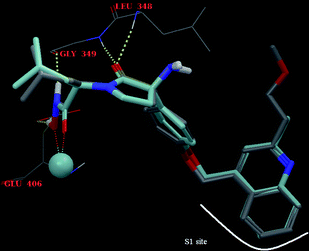 |
| | Fig. 3 H-bond interactions of compound 11p (elemental colour) and BMS-561392 (1) (turquoise colour). | |
As described earlier, the S1 site of TACE is larger and bend-shaped, with respect to most of the MMPs and ADAM-10, thus suitable substituents on the aromatic moiety are known to exhibit higher potency as well as good selectivity towards TACE over MMPs.21–23 As shown in Fig. 4, the methoxy-ethyl group on the 2nd position of the quinoline ring (11p) protrudes from a groove (termed the alkoxyalkyl pocket), which was not observed with 1. Together, the molecular docking study results indicate that the alkoxyalkyl substituents (compounds 11o and 11p) at the 2nd position of the quinoline ring system are essential for potent and selective TACE inhibitory activity profile.
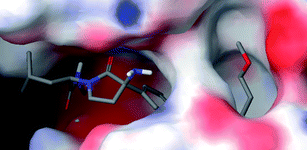 |
| | Fig. 4 Compound 11p docked pose in the active site of TACE with the alkoxyalkyl group at the 2nd position of the quinoline protruding from the alkoxyalkyl pocket. | |
In vivo TNF-α inhibition activity and pharmacokinetic (PK) studies
The in vivo TNF-α inhibition activity of most potent and selective compounds (11o–p) was assessed using LPS induced TNF-α production in Swiss Albino Mice (SAM).26 In this model, mice (n = 6) were dosed orally with different concentrations of test compounds, 0.5 h prior to the LPS (50 mg kg−1, iv) injection and the blood samples were drawn 1 h after LPS-stimulation.¶ The TNF-α levels in serum were determined using a TNF-α ELISA kit and ED50 values were determined. Compared to ex vivo TNF-α inhibition and in vitro TACE inhibitory activity results, significant difference in the in vivo TNF-α inhibitory activity were observed. Among the two test compounds (11o–p) screened in vivo, compound 11p was found to be around 2-fold more potent (ED50: 3.1 mg kg−1, po) than 11o (ED50: 7.2 mg kg−1, po) and standard compound 1 (ED50: 6.9 mg kg−1, po). In order to understand the pharmacokinetic (PK) profile of test compounds, a comparative single dose (5 mg kg−1; iv/po) PK study of our most potent compound (11p), along with standard compound 1 was carried out in male Wistar rats and the various PK parameters were recorded (Table 3). In a single dose PK study (5 mg kg−1, po), test compound 11p showed rapid tmax and clearance, good AUC and moderate half-life, while standard compound 1 showed extended tmax and t1/2 and moderate AUC and clearance. Compared to standard compound 1, test compound 11p showed 3-fold higher bioavailability (F: ∼60%). One of the reasons why compound 11p showed good in vivo TNF-α inhibitory activity could be correlated with its rapid tmax, good plasma exposure and higher bioavailability.
Table 3 Pharmacokinetic (PK) parameters comparison of compound 11p with 1 at 5 mg kg−1 (n = 9)
| |
PK parametersa |
Compd 11p |
Compd 1 |
|
Single dose (5 mg kg−1; iv/po) PK study for compound 11p and 1 was carried out in fasting male Wistar rats (n = 9) and plasma concentration of compounds were determined by LC–MS/MS, data represented as mean ± SD.
|
| iv |
t
1/2 (h) |
1.09 ± 0.1 |
2.69 ± 0.1 |
|
k
el (h−1) |
1.02 ± 0.1 |
0.30 ± 0.1 |
|
AUC (h ng ml−1) |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 726 ± 188 726 ± 188 |
32356 ± 899 |
| po |
t
max (h) |
0.33 ± 0.2 |
1.21 ± 0.01 |
|
t
1/2 (h) |
3.81 ± 0.2 |
8.67 ± 0.26 |
|
k
el (h−1) |
0.44 ± 0.01 |
0.08 ± 0.05 |
|
AUC (h ng ml−1) |
11776 ± 103 |
5970 ± 99 |
|
F (%) |
59.69% |
18.45% |
Safety pharmacology
To assess the comparative safety profile of compound 11p over standard compound 1, repeat dose acute toxicity studies (28 days) of both the compounds were carried out in male Wistar rats (100 mg kg−1, po, bid) and various parameters such as gross pathology, clinical signs, body weight, organ weights, and serum chemistry/hematological changes were recorded. In general, daily oral administration of compounds 11p and 1 did not affect the survival of Wistar rats and also no adverse changes related to gross pathology, clinical signs, body weight and feed consumption were noticed as compared to control group. However, compound 1 showed significant changes in relative organ weights and hematological parameters.
Some of the key parameters, such as comparative hematological changes (Table 4), and relative organ weights (Table 5), which are relevant to hepatotoxicity assessment are described in detailed. Acute hepatocellular injury markers such as, ALT (COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundalanine aminotransferase) and AST (aspartate aminotransferase) alone or in combination with hepatobiliary markers such as ALP (alkaline phosphatase) and total COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundbilirubin (TBILI) are primarily considered for the assessment of hepatotoxicity in non-clinical studies.34 As shown in Table 4, the hematological parameters (WBC and RBC) of compounds 1 and 11p were found to be comparable to that of control animals. Similarly, compound 11p showed no significant changes in serum ALP, AST, ALT and TBILI as compared to the control group. However, compound 1 treated group showed significantly elevated levels of all the serum liver enzymes (ALP, AST, ALT and TBILI), which infer its hepatotoxic effects in animal model. Also, compound 1 treated group elicit hepatocellular hypertrophy, marked by significant increased in liver weight as compared to control and compound 11p treated groups (Table 5), while other key organs (heart, kidney, spleen and brain) remained un-changed.
Table 4 Comparison hematological parameters and serum chemistry of compound 11p with 1a
| Parameters |
Compounds |
| Control |
11p
|
1
|
|
Values expressed as mean ± SD; n = 9, Male WR, dose 100 mg kg−1, po (bid), 28 days repeated dose toxicity study.
Represent significant elevation of serum liver enzymes at P < 0.01, compared to control.
|
|
WBC (103 μl−1) |
8.20 ± 0.33 |
8.41 ± 0.21 |
8.99 ± 0.41 |
|
RBC (106 μl−1) |
7.38 ± 0.11 |
8.01 ± 0.23 |
7.69 ± 0.99 |
| ALP (U L−1) |
134.66 ± 5.3 |
121.2 ± 12.1 |
523.43 ± 7.1b |
|
AST (U L−1) |
147.16 ± 11.75 |
140.2 ± 9.32 |
457.21 ± 8.3b |
| ALT (U L−1) |
20.78 ± 1.31 |
21.04 ± 8.36 |
39.22 ± 1.99b |
|
TBILI (mg dL−1) |
0.14 ± 0.01 |
0.17 ± 0.08 |
0.79 ± 0.02b |
Table 5 Comparison of relative organ weights (%) after 28 days repeat dose treatment with compound 11p with 1a
| Organs |
compounds |
| Control (Vehicle) |
11p (100 mg kg−1, po, bid) |
1 (100 mg kg−1, po, bid) |
|
Values expressed as mean ± SD; n = 9, Male WR, dose 100 mg kg−1, po, 28 days repeated dose toxicity study.
Significant difference from control at 5% level (p < 0.05).
|
| Heart |
0.345 ± 0.007 |
0.358 ± 0.008 |
0.351 ± 0.021 |
| Liver |
3.659 ± 0.1 |
3.801 ± 0.069 |
4.993 ± 0.127b |
| Kidney |
0.821 ± 0.03 |
0.878 ± 0.024 |
0.831 ± 0.04 |
| Spleen |
0.201 ± 0.007 |
0.203 ± 0.01 |
0.211 ± 0.026 |
| Brain |
0.727 ± 0.024 |
0.703 ± 0.028 |
0.714 ± 0.015 |
In summary, ex vivo, in vitro and molecular docking study results clearly demonstrated that the potency and selectivity of γ-lactam hydroxamate based TACE inhibitors can be modulated using suitable substituents at the 2nd position of the quinoline ring system. Furthermore, it was observed that suitable substituents at the 2nd position contributed significantly towards improvement in the in vivo TNF-α inhibition activity, which could be correlated with an improved oral bioavailability. Finally, in repeat dose acute toxicity study, the most potent and selective test compound 11p showed no adverse changes related to gross pathology, clinical signs and liver toxicity, indicating that the good selectivity profile of new class γ-lactam hydroxamate based TACE inhibitors over MMPs and ADAM-10 is essential to overcome hepatotoxicity concerns associated with similar class of TACE inhibitors. Overall, these results demonstrated discovery of a new class of highly potent and selective γ-lactam hydroxamate based TACE inhibitors which may be clinically useful for the safe and effective treatment of various inflammatory conditions.
Acknowledgements
We are grateful to the management of Zydus Group for encouragement and thankful to the analytical department for analytical support.
References
- M. A. Palladino, Nat. Rev. Drug Discovery, 2003, 2, 736–746 CrossRef CAS.
- R. A. Black, C. T. Rauch, C. J. Kozlosky, J. J. Peschon, J. L. Slack, M. F. Wolfson, B. J. Castner, K. L. Stocking, P. Reddy, S. Srinivasan, N. Nelson, N. Bolani, K. A. Schooley, M. Gerhart, R. Devis, J. N. Fitzner, R. S. Johnson, R. J. Paxton, C. J. March and D. P. Cerretti, Nature, 1997, 385, 729–732 CrossRef CAS.
- M. L. Moss, S. L. Jin, M. E. Milla, W. Burkhart, H. L. Carter, W. J. Chen, W. C. Clay, J. R. Didsbury, D. Hassler, C. R. Hoffman, T. A. Kost, M. H. Lambert, M. A. Leesnitzer, P. McCauley, G. McGeehan, J. Mitchell, M. Moyer, G. Pahel, W. Rocque, L. K. Overton, F. Schoenen, T. Seaton, J.-L. Su, J. Warner, D. Willard and J. D. Becherer, Nature, 1997, 385, 733–736 Search PubMed.
- M. Feldmann, F. M. Brennan and R. N. Maini, Annu. Rev. Immunol., 1996, 14, 397–440 CrossRef CAS.
- M. Odeh, Clin. Immunol. Immunopathol., 1997, 83, 103–116 Search PubMed.
- D. R. Bertolini, G. E. Nedwin, T. S. Bringman, D. D. Smith and G. R. Mundy, Nature, 1986, 319, 516–518 Search PubMed.
- M. H. Rabinowitz, R. C. Andrews, J. D. Becherer, D. M. Bickett, D. G. Bubacz, J. G. Conway, D. J. Cowan, M. Gaul, K. Glennon, M. H. Lambert, M. A. Leesnitzer, D. L. McDougald, M. L. Moss, D. L. Musso and M. C. Rizzolio, J. Med. Chem., 2001, 44, 4252–4267 CrossRef CAS.
- J. J. W. Duan, L. Chen, Z. R. Wasserman, Z. Lu, R. Liu, M. B. Covington, M. Qian, K. Hardman, R. L. Magolda, R. C. Newton, D. D. Christ, R. R. Wexler and C. P. Decicco, J. Med. Chem., 2002, 45, 4954–4957 Search PubMed.
- J. J. W. Duan, M. E. Voss, L. Chen, W. Li, D. T. Meyer, Z. R. Wasserman, K. D. Hardman, R. Liu, M. B. Covington, M. Qian, S. Mandlekar, D. D. Christ, J. M. Trzaskos, R. C. Newton, R. L. Magolda, R. R. Wexler and C. P. Decicco, Bioorg. Med. Chem. Lett., 2003, 13, 2035–2040 CrossRef CAS.
- G. T. Le and G. Abbenante, Curr. Med. Chem., 2005, 12, 2963–2977 CrossRef CAS.
- S. Wojtowicz-Praga, J. Torri, M. Johnson, V. Steen, J. Marshall, E. Ness, R. Dickson, M. Sale, H. S. Rasmussen, T. A. Chiodo and M. J. Hawkins, J. Clin. Oncol., 1998, 16, 2150–2165 Search PubMed.
- C. B. Xue, X. He, R. L. Corbett, J. Roderick, Z. R. Wasserman, R.-Q. Liu, B. D. Jaffee, M. B. Covington, M. Qian, J. M. Trzaskos, R. C. Newton, R. L. Magolda, R. R. Wexler and C. P. Decicco, J. Med. Chem., 2001, 44, 3351–3354 Search PubMed.
- M. L. Moss, L. S. Tavron and R. Nedelman, Nat. Clin. Pract. Rheumatol., 2008, 4, 300–309 Search PubMed.
- M. M. Thabet and T. W. Huizinga, J. Curr. Opin. Invest. Drugs, 2006, 7, 1014–1019 Search PubMed.
- M. L. Moss, J. M. White, M. H. Lambert and R. C. Andrews, Drug Discovery Today, 2001, 6, 417–426 CrossRef CAS.
- Z. Zhu, R. Mazzola, L. Sinning, B. McKittrick, X. Niu, D. Lundell, J. Sun, P. Orth, Z. Guo, V. Madison, R. Ingram and B. M. Beyer, J. Med. Chem., 2008, 51, 725–725 Search PubMed.
- D. Georgiadis and A. Yiotakis, Bioorg. Med. Chem., 2008, 16, 8781–8794 CrossRef CAS.
- K. Reiss and P. Saftig, Semin. Cell Dev. Biol., 2009, 20, 126–137 Search PubMed.
- Y. Zhang, M. Hegen, J. Xu, J. C. Keith, G. Jin, X. Du, T. Cummons, B. J. Sheppard, L. Sun, Y. Zhu, V. R. Rao, Q. Wang, W. Xu, R. Cowling, C. L. Nickerson-Nutter, J. Gibbons, J. Skotnicki, L. L. Lin and J. Levin, Int. Immunopharmacol., 2004, 4, 1845–1857 Search PubMed.
- J. J. W. Duan, L. Chen, Z. R. Wasserman, Z. Lu, R. Q. Liu, M. B. Covington, M. Qian, K. D. Hardman, R. L. Magolda, R. C. Newton, D. D. Christ, R. R. Wexler and C. P. Decicco, J. Med. Chem., 2002, 45, 4954–4957 Search PubMed.
- X. Niu, S. Umland, R. Ingram, B. M. Beyer, Y. H. Liu, J. Sun, D. Lundell and P. Orth, Arch. Biochem. Biophys., 2006, 451, 43–50 Search PubMed.
- V. Lukacova, Y. Zhang, D. M. Kroll, S. Raha, D. Comez and S. Balaz, J. Med. Chem., 2005, 48, 2361–2370 Search PubMed.
- Z. R. Wasserman, J. J. Duan, M. E. Voss, C. B. Xue, R. J. Cherney, D. J. Nelson, K. D. Hardman and C. P. Decicco, Chem. Biol., 2003, 10, 215–223 Search PubMed.
- Z. Zhu, R. Mazzola, L. Sinning, B. McKittrick, X. Niu, D. Lundell, J. Sun, P. Orth, Z. Guo and V. Madison, J. Med. Chem., 2008, 51, 725–736 Search PubMed.
- A. Huang, D. Joseph-McCarthy, F. Lovering, L. Sun, W. Wang, W. Xu, Y. Zhu, J. Cui, Y. Zhang and J. I. Levin, Bioorg. Med. Chem., 2007, 15, 6170–6181 Search PubMed.
- J. I. Levin, J. M. Chen, K. Cheung, D. Cole, C. Crago, E. D. Santos, X. Du, G. Khafizova, G. MacEwan, C. Niu, E. J. Salaski, A. Zask, T. Cummons, A. Sung, J. Xu, Y. Zhang, W. Xu, S. Ayral-Kaloustian, G. Jin, R. Cowling, D. Barone, K. M. Mohler, R. A Black and J. S. Skotnicki, Bioorg. Med. Chem. Lett., 2003, 13, 2799–2803 Search PubMed.
- Z. R. Wasserman, J. J. Duan, M. E. Voss, C. B. Xue, R. J. Cherney, D. J. Nelson, K. D. Hardman and C. P. Decicco, Chem. Biol., 2003, 10, 215–223 Search PubMed.
-
B. B. Lohray, V. B. Lohray, M. R. Jain and P. S. Thombare, WO 2005077937.
- E. S. H. El Ashray, E. S. Ramadana, H. A. Hamida and M. Hagara, Synth. Commun., 2005, 35, 2243–2250 Search PubMed.
-
R. E. Waltermire, S. A. Savage, S. Campagna, N. A. Magnus, P. N. Confalone, M. Yates and D. J. Meloni, WO 2003104220.
- N. Xiaoda, U. Shelby, I. Richard, M. B. Brian, H. L. Yan, S. Jing, L. Daniel and O. Peter, Arch. Biochem. Biophys., 2006, 451, 43–50 Search PubMed.
-
Glide 5.0; Schrodinger, LLC: New York, NY, 2008 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- L. Boone, D. Meyer, P. Cusick, D. Ennulat, A. P. Bolliger, N. Everds, V. Meador, G. Elliott, D. Honor, D. Bounous and H. Jordan, Vet. Clin. Pathol., 2005, 34, 182–188 Search PubMed.
Footnotes |
| † ZRC communication No: 326 (Part of PhD work of Mr. A. Argade) |
| ‡ The authors have declared no conflict of interest. |
| § Electronic supplementary information (ESI) available: Experimental data, materials and methods for in vitro and in vivo assay. See DOI: 10.1039/c0md00261e |
| ¶ All the animal experiments were conducted according to the internationally valid guidelines following approval by the ‘Zydus Research Center Animal Ethical Committee’. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.