Structure-based design, synthesis, and profiling of potent and selective neuronal nitric oxide synthase (nNOS) inhibitors with an amidinothiophene hydroxypiperidine scaffold
Guyan
Liang
*a,
Kent
Neuenschwander
b,
Xin
Chen
a,
Linli
Wei
b,
Randall
Munson
b,
Gerardo
Francisco
a,
Anthony
Scotese
b,
Gregory
Shutske
b,
Mark
Black
c,
Sharkir
Sarhan
c,
Jason
Jiang
d,
Isabelle
Morize
a and
Roy J.
Vaz
a
aDepartment of Chemical and Analytical Science, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA. E-mail: guyan.liang@sanofi-aventis.com; Tel: +1-908-231-4573
bDepartment of CNS Medicinal Chemistry, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
cDepartment of CNS Pharmacology, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
dDepartment of DMPK-S, Sanofi-Aventis Pharmaceuticals, Route 202-206, Bridgewater, NJ 08807, USA
First published on 26th January 2011
Abstract
A novel series of nNOS inhibitors with an amidinothiophene-hydroxypiperidine scaffold was designed based on X-ray structures and in-silico models. Three classes of inhibitors with this scaffold were synthesized and tested for their nNOS activity and eNOS selectivity. Compounds with a linear aliphatic amine linker demonstrated a superior property over those with a sulfonamide or an amide-like linker.
Nitric oxide (NO), an essential signaling molecule and a ubiquitous biological messenger, is involved in various physiological processes in human and exerts a variety of regulatory and cytostatic functions. Nitric oxide is synthesized by Nitric Oxide Synthases (NOS, EC 1.14.13.39), which catalyze the oxidation of L-arginine to L-citrulline and NO in a NADPH and O2 dependent process. Three isoforms of NOS have been identified, each of which is associated with a distinct physiological function. Two isoforms, neuronal NOS (nNOS) and endothelial NOS (eNOS), are constitutively expressed and intermittently produce small amounts of NO, while the third isoform, iNOS, is inducible by cytokines for both cytoprotective and cytotoxic effects. Neuronal NOS, along with two other isoforms, has been widely studied and its association with variety of illnesses has been well recognized, including but not limited to neurondegeneration during stroke,1 spinal transmission of pain,2 migraine headaches,3 Parkinson's disease,4 Alzheimer's disease,5 and schizophrenia.6 A potent and selective nNOS inhibitor with a proper ADMET profile is of significant therapeutic interest.
As a therapeutic target, nNOS has been an active area for both academic and industrial researchers. Inhibitors of nNOS with various structural scaffolds, as well as different selectivity and ADMET profiles, have been published, including peptidomimetics,7H4B derivatives,8 and other non-peptidomimetic compounds.9 In the present study, a group of compounds featuring a hydroxypiperidine moiety is reported, structure–activity relationship (SAR) among them is established, and critical interactions between nNOS and inhibitors are identified.
To the advantage of this project, X-ray structures of all three NOS isoforms were available from either public domain or our in-house Structural Biology efforts. Bearing high similarity to eNOS and iNOS, nNOS is a multi-domain enzyme, which can be divided into the N-terminus catalytic domain, the C-terminus reductase domain, and the calmodulin (CaM) binding domain that bridges the catalytic domain and the reductase domain. While the reductase domain contains flavin mononucleotide (FMN) and the reduced NADP, both of which are critical for the electron transfer and catalytic function of nNOS, our structure-based design effort was largely focused on the catalytic domain, especially around the HEME containing catalytic center (substrate binding pocket) and the nearby cofactor, tetrahydrobiopterine (H4B).
In general, all three isoforms of NOS exhibit a dimeric fold and have a wide-open channel connecting the HEME-containing catalytic site to the protein surface. This channel is solvent exposed and offers easy access to the catalytic site for the natural substrate, L-arginine. The substrate binding pocket, located right above the HEME ring, is a dome-shaped pocket. As shown in Fig. 1, the residues around the substrate binding pocket present a rich collection of interaction features which offers great potential for structure-based inhibitor design. On the right hand side, the α and β carbon atoms of S585, along with the side chain atoms of F584 and M570, form a hydrophobic pocket above the HEME. On the left hand side, the backbone carbonyl oxygen of W587 and the side chain of E592 present a critical H-bonding network, which plays a pivotal role in the substrate and inhibitor binding. The binding contribution of both the H-bonding network and the hydrophobic pocket are generic to all three isoforms of NOS and are not specific to nNOS. Residues in this region are identical among all three isoforms and their X-ray structures superimpose well on top of one another.
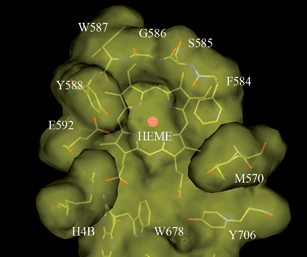 | ||
| Fig. 1 A top-down view of the substrate binding pocket of rat nNOS. This picture is generated based on an in-house X-ray structure with molecular modeling software InsightII® from Accelrys®. | ||
Also common to all three isoforms of NOS is the entrance to the substrate binding pocket which is featured by two propionic acids and the cofactor, H4B. This region is highly polarized and offers a good opportunity for a charge-charge interaction or an H-bonding interaction with the enzyme. However, because all those interactions are generic to all three isoforms, any binding affinity gained over those interactions most likely will not help to enhance selectivity. Good selectivity of nNOS inhibitors, especially against eNOS is critically important to minimize undesirable side effects due to the ubiquitous expression of eNOS.
The highly conserved binding features among the three isoforms posed a major challenge to the development of selective nNOS inhibitors. Based on a rich collection of proprietary iNOS and nNOS X-ray structures co-crystallized with our own inhibitors and publically available eNOS X-ray structures, such as 1P6L, 1P6M, 1P6N, and 1P6O,10 a “selectivity modulating region” was identified. This region, consisting of hydrophobic residues L337, M570, W678, and Y706, is located approximately 15 Å away from the center of the HEME ring and 8 Å from the nearest propionic acid. In this region, residues of the superimposed X-ray structures of three isoforms deviate more significantly than those in the region closer to the HEME ring. Additionally, in molecular dynamics (MD) simulations, residues in this region exhibit more distinguishable folding flexibility pattern. Therefore, it is presumably possible to introduce protein-ligand interactions that are differentiable between nNOS and other NOS isoforms, which could lead to an enhanced selectivity. Following this strategy, inhibitors with an amidinothiophene hydroxypiperidine scaffold and various linkers were designed to project a part of the inhibitor to the selectivity modulating region to leverage on the unique hydrophobic interaction with residues in the area.
As shown in Fig. 2, the phenyl-amidinothiophene group of the inhibitor binds right above the HEME ring with the thiophene ring tucked between the HEME ring and the backbone of S585 and G586. This binding mode is further stabilized by an H-bonding network between the amidino group of the inhibitor and between the backbone carbonyl oxygen of W587 and the carboxyl oxygen of the E592 side chain. The hydroxypiperidine of the inhibitor, sitting right above the two propionic acids of the HEME group, has two possible binding modes based on docking studies using Glide. In the energetically preferred binding mode based on Glide score, as shown in Fig. 2, the piperidine ring stays perpendicular to the phenyl ring of the scaffold and the HEME ring of nNOS. The hydroxypiperidine can also adapt an alternative binding mode and stay nearly parallel to the phenyl ring with the hydroxyl group facing down toward the two propionic acids. While both binding modes were evidenced by X-ray structures and explored during the chemical optimization, the binding mode shown in Fig. 2 was seen more often in X-ray structures and remained as our main focus during the compound optimization.
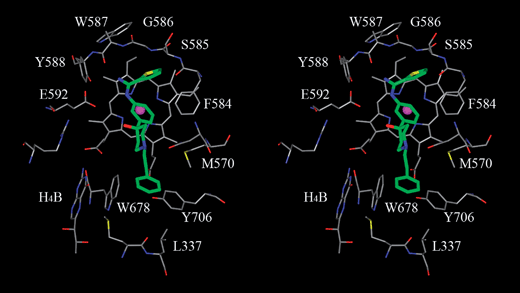 | ||
| Fig. 2 A stereo view of a modeled inhibitor with the amidinothiophene hydroxypiperidine scaffold (8a without the chloro substitution) in the rat nNOS substrate binding pocket. This modeled binding mode was refined with molecular modeling software InsightII®/Discover® from Accelrys® based on docking poses generated by Glide® from Schrodinger®. This binding mode was later confirmed by an in-house X-ray structure. Carbon atoms of nNOS are colored in gray and carbon atoms of the modeled inhibitor is colored in green. | ||
The hydroxypiperidine ring binds in the channel connecting the substrate binding pocket and the protein surface. While the area in which the amidinothiophene ring (referred to as the “head”) binds is limited in space by the dome-shaped pocket, another end of the inhibitor (referred to as the “tail”) sits in a much more spacious area, which can accommodate a variety of different functional groups. In the present study, we focused our effort largely on the chemical optimization of the tail and the exploration of various linker groups connecting the tail to the hydroxypiperidine ring and further to the head. During the course of the project, three linker groups were explored, including amide-like, sulfonamide, and simple aliphatic amines.
Based on X-ray structures and in-silico models, two major factors may influence the binding affinity of those nNOS inhibitors. The first factor is the steric hinderance and second factor is the charge-charge interaction between the linker group and the two propionic acids. Sulfonamide as a linker group may cause a moderate steric hinderance which can be partially relieved by relaxing the side chains of nearby residues. Those changes, even if minor, still carry enthalpic penalty to overall binding affinity. This hypothesis is supported by several X-ray structures, in which inhibitors with a hydroxypiperidine-sulfonamide linker binds in a different orientation from those with non-sulfonamide linkers. In this new binding mode presented by sulfonamide inhibitors, several residues with direct interactions with the inhibitor shifted away from their energetically preferred positions, which was not observed for inhibitors with a non-sulfonamide linker.
The second factor which may impact the potency is shared by compounds with the sulfonamide linker and those with the amide-like linkers including carboxamide, urea, and carbamic acid. The linker part of those inhibitors binds in an area which is close to the two propionic acids of the HEME ring, where a positively charged group is preferred. With a sulfonamide or amide-like nitrogen atom instead of an aliphatic amino nitrogen, the charge-charge interaction between the inhibitor and the two propionic acids is weakened, which consequently decreases the binding affinity of the inhibitor.
Following this rationale, it was hypothesized that both issues could be avoided by replacing the sulfonamide or amide-like linker group with a linker containing only aliphatic amino nitrogen, which lessens the steric constrain in the region and preserves the critical charge-charge interaction with the two propionic acids. To confirm this rationale, representative compounds with all three linkers were synthesized following Scheme 1 with various R groups and tested for their nNOS potency.
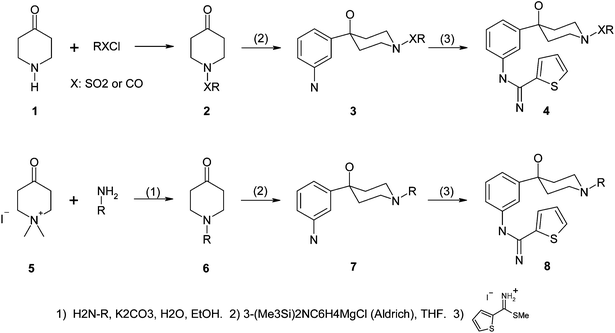 | ||
| Scheme 1 Synthetic schemes of amidinothiophene-hydroxypiperidine inhibitors. | ||
As shown in Scheme 1, piperidin-4-one 1 reacted with various RXCl such as alkylsulfonyl chloride in the presence of a base to give 2 in good yields. Compound 2 was treated with 3-[bis(trimethylsilyl)-amino]phenylmagnesium chloride (1M in THF, Aldrich) and then followed by aq. acidic workup (10% aq. HCl) to give 3. Compound 3 was heated with thiophene-2-carboximidothioic acid methyl ester HI salt11 in EtOH under refluxing condition to give 4. Alternatively, compound 5 was treated with corresponding amines under basic and refluxing condition to give 6 in high yields.12 Compound 6 was further converted to 8 by following the same procedure.
Inhibition against nNOS was measured by an enzymatic assay, which was based on the conversion of L-arginine to citrulline originally described by Bredt and Snyder.13 Recombinant rat nNOS was incubated in Costar polyethylene 96-well microplates for 30 min at 37 °C and pH 7.4 with 5.0 μM L-arginine, 50 mM HEPES, 15 μM H4B, 1 μM FAD, 100 μM DTT, 10 μg ml−1 calmodulin, 1 mM EDTA, 1.25 mM CaCl2, 1 mM NADPH, and the compound to be tested in a final volume of 90 μL. The incubation was terminated by transferring 60 μL aliquots of the assay solution into Millipore MAHV N45 plates packed with 80 μL Dowex AG cation exchange resin prewashed with 100 uL of 100 mM HEPES buffer of pH 5.5 containing 10 mM EGTA. After filtering under vacuum into opaque scintillation plates, 220 μL of Packard Microscint-40 were added to each well. The plates were counted on a Packard TopCount to quantify the radio-labeled citrulline product, based on which nNOS activity and compound inhabitation were derived.
As shown in Table 1, sulfonamide derivatives (4a and 4b) as a group, demonstrate weaker potency than other compounds, which is consistent with the structural observation that inhibitors with a hydroxypiperidine sulfonamide linker encounter a moderate steric hinderance by surrounding residues and that the charge-charge interaction with the two propionic acids was diminished due to a significantly decreased positive charge on the sulfonamide nitrogen comparing to aliphatic amines. Inhibitors with a urea linker, 4x, and a carbamic acid linker, 4y, exhibit an enhanced potency comparing to those with a sulfonamide linker but is less than compounds with a linker of aliphatic amines. This is also consistent with the protein-ligand interactions observed from both in-silico models and X-ray co-crystal structures, in which the charge-charge interaction with the two propionic acids is much stronger for compounds with an aliphatic amine linker than those with a sulfonamide or an amide-like group.
|
|
|||
|---|---|---|---|
| Cmpd | X | R | IC50 (nM) |
| 4a | –SO2– | –(CH2)2CH3 | 706 |
| 4b | –SO2– | –CH2(CH3)2 | 551 |
| 4x | –(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)– O)– |
–N(CH2)3CH3 | 694 |
| 4y | –(CO)– |
–OCH2C6H5 | 376 |
| 8a | –(CH2)2– | -m-chlorophenyl | 131 |
| 8b | –(CH2)2– | -o, m-dichlorophenyl | 129 |
| 8c | –(CH2)2– | -m, p-dichlorophenyl | 17 |
One major objective of this chemical optimization was to identify proper tails that exhibit divergent SARs between nNOS and eNOS activities so that the selectivity against eNOS can be optimized further. Following our structure-based strategy to explore the selectivity modulating region, tail groups with various sizes and interaction features were coupled with the amidinothiophene hydroxypiperidine scaffold and an aliphatic amine linker. As shown by compounds 8a, 8b, and 8c in Table 2, inhibitors with a chlorophenyl tail exhibit good potency for nNOS and satisfactory selectivity against eNOS with many of them pushing the detection limit of the eNOS assay.
| Cmpd | nNOS (nM) | eNOS (μM) | Selectivity ratio | Total metabolism |
|---|---|---|---|---|
| 4a | 706 | 18.2 | 26 | 25% |
| 4b | 551 | 23.0 | 42 | 16% |
| 4x | 694 | 10.0 | 14 | 54% |
| 4y | 376 | 60.1 | 159 | 77% |
| 8a | 131 | 57.5 | 438 | 39% |
| 8b | 129 | 100.0 | 775 | 23% |
| 8c | 17 | 28.3 | 1664 | 32% |
An in vitro human liver microsome assay was used as the first-line evaluation for metabolic stability. Compounds were incubated with purified human liver microsome for 20 min. At stoppage, the amount of compound remaining was measured from which the percent metabolism was calculated. As shown in Table 2, while inhibitors with an amide-like linker have medium to low metabolic stability, compounds with a sulfonamide linker or an aliphatic amine linker are reasonable stable.
In conclusion, three classes of compounds with an amidinothiophene hydroxypiperidine scaffold were designed, synthesized, and tested for their nNOS activity, eNOS selectivity, and human liver microsomal stability. While the limited number of representative compounds in each class is not large enough to warrant a broadly conclusive SAR, the potency comparison of those representative tool compounds is consistent with their interaction signatures observed in X-ray structures and in silico models. Compounds with a linear aliphatic amine linker exhibited a more favorable interaction with the protein in the linker region by eliminating the charge-charge repulsion and steric collision with surrounding residues. A “selectivity modulating region” represented by L337, M570, W678, and Y706 was identified.14 While this binding region prefers a hydrophobic group in general, inhibitors with a chlorophenyl group bound in this region have shown superior selectivity against eNOS. Considering all properties together including the metabolic stability from the human liver microsome assay, inhibitors with an amidinothiophene hydroxypiperidine scaffold, an aliphatic amine linker, and a chlorophenyl tail have the overall best profile.
References
- N. Toda, K. Ayajiki and T. Okamura, Pharmacol. Rev., 2009, 61, 62 CrossRef CAS.
- M. Puglia, G. Attaguile, F. Valenti and G. Pennisi, Recent Developments in Pain Research., 2005, 313 Search PubMed.
- P. D. Lokhande, B. S. Kuchekar, A. R. Chabukswar and S. C. Jagdale, Asian J. Biochem., 2006, 1, 1 Search PubMed.
- K. Aquilano, S. Baldelli, G. Rotilio and M. R. Ciriolo, Neurochem. Res., 2008, 33, 2416 CrossRef CAS.
- T. Malinski, Journal of Alzheimer's Disease, 2007, 11, 207 Search PubMed.
- A. Reif, S. Herterich, A. Strobel, A.-C. Ehlis, D. Saur, C. P. Jacob, T. Wienker, T. Toepner, S. Fritzen, U. Walter, A. Schmitt, A. J. Fallgatter and K.-P. Lesch, Molecular Psychiatry, 2006, 11, 286 Search PubMed.
- (a) J. Seo, J. Igarashi, H. Li, P. Martasek, L. J. Roman, T. L. Poulos and R. B. Silverman, J. Med. Chem., 2007, 50, 2089 CrossRef CAS; (b) H. Ji, J. A. Gomez-Vidal, P. Martasek, L. J. Roman and R. B. Silverman, J. Med. Chem., 2006, 49, 6254 CrossRef CAS; (c) J. A. Gomez-Vidal, P. Martasek, L. J. Roman and R. B. Silverman, J. Med. Chem., 2004, 47, 703 CrossRef CAS; (d) J.-M. Hah, P. Martasek, L. J. Roman and R. B. Silverman, J. Med. Chem., 2003, 46, 1661 CrossRef CAS; (e) J.-Mi Hah, L. J. Roman, P. Martasek and R. B. Silverman, J. Med. Chem., 2001, 44, 2667 CrossRef CAS; (f) H. Huang, P. Martasek, L. J. Roman, B. S. S. Masters and R. B. Silverman, J. Med. Chem., 1999, 42, 3147 CrossRef CAS; (g) M. Cowart, E. A. Kowaluk, J. F. Daanen, K. L. Kohlhaas, K. M. Alexander, F. L. Wagenaar and J. F. Kerwin, Jr., J. Med. Chem., 1998, 41, 2636 CrossRef CAS; (h) H. Q. Zhang, W. Fast, M. A. Marletta, P. Martasek and R. B. Silverman, J. Med. Chem., 1997, 40, 3869 CrossRef CAS; (i) R. B. Silverman, H. Huang, M. A. Marletta and P. Martasek, J. Med. Chem., 1997, 40, 2813 CrossRef CAS; (j) W. Fast, M. E. Huff and R. B. Silverman, Bioorg. Med. Chem. Lett., 1997, 7, 1449 CrossRef CAS; (k) J.-M. Hah, L. J. Roman and R. B. Silverman, Bioorg. Med. Chem., 2000, 8, 1931 CrossRef CAS; (l) B. N. A. Mbadugha, J. Seo, H. Ji, P. Martasek, L. J. Roman, T. M. Shea, H. Li, T. L. Poulos and R. B. Silverman, Bioorg. Med. Chem., 2006, 14, 3681 CrossRef CAS; (m) J. Seo, P. Martasek, L. J. Roman and R. B. Silverman, Bioorg. Med. Chem., 2007, 15, 1928 CrossRef CAS.
- L. G. Froehlich, P. Kotsonis, H. Traub, S. Taghavi-Moghadam, N. Al-Masoudi, H. Hofmann, H. Strobel, H. Matter, W. Pfleiderer and H. H. H. W. Schmidt, J. Med. Chem., 1999, 42, 4108 CrossRef CAS.
- (a) J. A. Lowe, III, W. Qian, S. E. Drozda, R. A. Volkmann, D. Nason, R. B. Nelson, C. Nolan, D. Liston, K. Ward, S. Faraci, K. Verdries, P. Seymour, M. Majchrzak, A. Villalobos and W. F. White, J. Med. Chem., 2004, 47, 1575 CrossRef CAS; (b) V. Collot, O. Sopkova-De, J. Santos, P. Schumann-Bard, N. Colloc'h, E. T. Mackenzie and S. Rault, J. Enzyme Inhib. Med. Chem., 2003, 18, 195 Search PubMed; (c) J. L. Collins, B. G. Shearer, J. A. Oplinger, S. Lee, E. P. Garvey, M. Salter, C. Duffy, T. C. Burnette and E. S. Furfine, J. Med. Chem., 1998, 41, 2858 CrossRef CAS; (d) A. Palumbo and A. Napolitano, Bioorg. Med. Chem. Lett., 2002, 12, 13 CrossRef CAS; (e) J. A. Lowe, III, W. Qian, R. A. Volkmann, S. Heck, J. Nowakowski, R. Nelson, C. Nolan, D. Liston, K. Ward, S. Zorn, C. Johnson, M. Vanase, W. S. Faraci, K. A. Verdries, J. Baxter, S. Doran, M. Sanders, M. Ashton, P. Whittle and M. Stefaniak, Bioorg. Med. Chem. Lett., 1999, 9, 2569 CrossRef; (f) P. Schumann, V. Collot, Y. Hommet, W. Gsell, F. Dauphin, J. Sopkova, E. T. MacKenzie, D. Duval, M. Boulouard and S. Rault, Bioorg. Med. Chem. Lett., 2001, 11, 1153 CrossRef CAS; (g) A. Palumbo and A. Napolitano, Bioorg. Med. Chem. Lett., 2002, 12, 13 CrossRef CAS; (h) S. Auvin, M. Auguet, E. Navet, J. J. Harnett, I. Viossat, J. Schulz, D. Bigg and P.-E. Chabrier, Bioorg. Med. Chem. Lett., 2003, 13, 209 CrossRef CAS; (i) D. M. Nason, S. D. Heck, M. S. Bodenstein, J. A. Lowe, R. B. Nelson, D. R. Liston, C. E. Nolan, L. F. Lanyon, K. M. Ward and R. A. Volkmann, Bioorg. Med. Chem. Lett., 2004, 14, 4511 CrossRef CAS; (j) J. Patman, N. Bhardwaj, J. Ramnauth, S. C. Annedi, P. Renton, S. P. Maddaford, S. Rakhit and J. S. Andrews, Bioorg. Med. Chem. Lett., 2007, 17, 2540 CrossRef CAS; (k) E. P. Erdal, P. Martasek, L. J. Roman and R. B. Silverman, Bioorg. Med. Chem., 2007, 15, 6096 CrossRef CAS; (l) G. R. Lawton, H. R. Ranaivo, L. K. Chico, H. Ji, F. Xue, P. Martasek, L. J. Roman, W. D. Martin and R. B. Silverman, Bioorg. Med. Chem., 2009, 17, 2371 CrossRef CAS; (m) R. B. Silverman, Acc. Chem. Res., 2009, 42, 439 CrossRef CAS.
- M. L. Flinspach, H. Li, J. Jamal, W. Yang, H. Huang, J. M. Hah, J. A. Gomez-Vidal, E. A. Litzinger, R. B. Silverman and T. L. Poulos, Nat. Struct. Mol. Biol., 2003, 11, 54.
- (a) A. Manaka and M. Sato, Synth. Commun., 2005, 35, 761 CrossRef CAS; (b) R. J. Gentile; R. J. Murray; J. E. MacDonarldW. C. Sharkespeare WO9505363, 1995.
- M. E. Kuehne and R. S. Muth, J. Org. Chem., 1991, 56, 2701 CrossRef CAS.
- D. S. Bredt and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 682.
- After this manuscript was submitted, the authors gladly recognized that Xue, Silverman, and co-workers reached a similar conclusion about the selectivity modulating region, although defined differently, in one of their latest publications, F. Xue, H. Li, J. Fang, L. J. Roman, P. Martasek, T. L. Poulos and R. B. Silverman, Bioorg. Med. Chem. Lett., 2010, 20, 6258 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |