Carba-LNA modified siRNAs targeting HIV-1 TAR region downregulate HIV-1 replication successfully with enhanced potency†
Suman
Dutta
a,
Nipa
Bhaduri
a,
Neha
Rastogi
a,
Sunita G.
Chandel
a,
Jaya Kishore
Vandavasi
b,
Ram Shankar
Upadhayaya
b and
Jyoti
Chattopadhyaya
*c
aInstitute of Molecular Medicine, BIPL, Building – B, Ist Floor, Block – EP & GP, Salt Lake Electronics Complex, Sector –V, Kolkata, 700 091, India
bInstitute of Molecular Medicine, International Biotech Park, Genesis Campus, Phase II Opp Infosys, Tal Mulshi Hinjewadi, Pune, 411 057, India
cProgram of Chemical Biology, Institute of Cell and Molecular Biology, Box 581, Biomedical Centre, Uppsala University, SE-75123, Uppsala, Sweden. E-mail: jyoti@boc.uu.se; Fax: +46-18-554495; Tel: +46-18-4714577
First published on 27th January 2011
Abstract
The conformationally-locked carbocyclic nucleosides carbaLNA (“jcLNA”) (Gagnon et al., Biochemistry, 2010, 49, 10166; Srivastava et al., J. Am. Chem. Soc., 2007, 129, 8362; Xu et al., J. Org. Chem., 2009, 74, 6534; Zhou and Chattopadhyaya, J. Org. Chem., 2010, 75, 2341; Zhou et al., J. Org. Chem., 2009, 74, 118) are chemically engineered by fusing a carbocyclic ring at the C2′ to C4′ chiral centres in a stereospecific manner at the α-face of the pentose-sugar of the native nucleosides. The benefit of the chemically-modified oligonucleotides with the jcLNA scaffold has been shown to be their uniquely enhanced nuclease resistance in the blood serum as well as their improved RNase H recruitment capability to cleave the target RNA in the hybrid antisense-RNA duplex when used as an antisense agent, compared to those of locked nucleic acid (LNA) modified counterparts. Herein we report the relative inhibition efficiency of HIV-1 by jcLNA modified siRNAs targeting TAR region compared to those of the LNA counterparts, in that the former were found to exhibit improved silencing efficiency and displayed enhanced stability in human serum with negligible cytotoxicity compared to those of the latter. A single jcLNA substitution as the 3′-overhang of the guide strand displayed near native-like IC50 value (of 4.01 ± 0.87 nM compared to the nearly two-fold higher IC50 value of 7.15 ± 1.57 nM for LNA modified counterparts, and of the native siRNA of 1.84 ± 0.16 nM) and significantly higher t1/2 value for the stability in serum (11.9 h for jcLNA, 6.8 h for LNA and 3.0 h for native), thereby showing that the efficiency of jcLNA-modified-siRNAs is supported by stability without compromising the native-like efficiency and target RNA recognition and subsequent down-regulation. Amongst all the modified siRNAs so far used to target HIV-1 TAR region, the best IC50 value was obtained for the doubly-modified siRNA in which jcLNA substitution was introduced both at position 1 and 20 of the antisense strand (T1 + T20, i.e. jcLNA11 which showed IC50 value of 0.54 ± 0.14 nM). The IC50 of this doubly-modified siRNA was more than three-fold lower than that of the native and two-fold lower than that of LNA modified counterpart, i.e. LNA12: IC50: 1.13 ± 0.27 nM. Hence the strategy to chemically modify the native siRNAs by substitution with the jcLNA can be considered as a significant development, leading to both enhanced siRNA efficiency and serum stability over that of the native.
Introduction
RNA interference (RNAi) is mediated by small interfering RNA (siRNA) duplexes of about 21 nucleotide lengths, which work through RNA-Induced Silencing Complex (RISC) and direct the cleavage of sequence specific cellular target mRNA.6 This has been successfully used to suppress the expression of transfected and endogenous genes in a large number of mammalian cells.7siRNAs in their native form, however present several problems as potential therapeutic tools including nuclease degradation, off-targeting, delivery and poor tissue distribution.8 These shortcomings have triggered the development of chemically-modified nucleotide analogues which are aimed to give better stability and nuclease protection over the native counterparts.9–11 Amongst an array of modified nucleotides developed12–15 LNA and its modifications16–22 have emerged as credible alternatives. The nuclease resistance and also the efficacy of LNA modification in gene silencing has been described.23–25 In a promising development4 jcLNA modified nucleosides had been shown to present remarkably enhanced stability over LNA. The structural difference between jcLNA nucleoside (Fig. 1a) and LNA nucleoside (Fig. 1b) lies in the fact that –2′-O– in LNA (Fig. 1b) is replaced with a –CH(CH3)– group in the fused alkane chain in jcLNA (Fig. 1a).
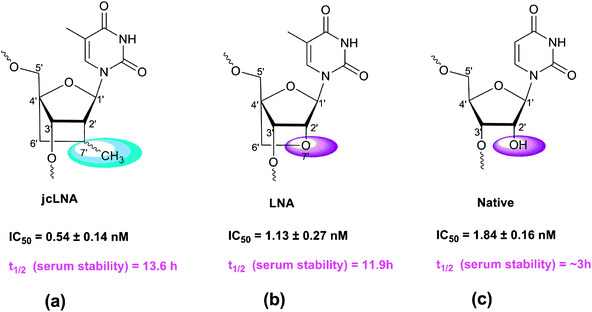 | ||
| Fig. 1 Structures of jcLNA-T or LNA-T modified nucleotides and that of the native nucleotide. | ||
The C4′–CH2–CH (Me)–C2′ alkane bridge in jcLNA, as shown in Fig. 1a, is covalently linked to the C-2′ and C-4′ chiral centers of the β-D-pentofuranose unit. This carba-bridge in jcLNA on the α-face of the sugar unit, as shown in Fig. 1b, reduces the conformational flexibility of the ribose sugar to the locked 3′-endo (North-type) conformation,26 as the oxa-bridge in LNA, as shown in Fig. 1b, thereby reducing the entropic penalty for hybridization with the target RNA in both cases. What makes the jcLNA however unique is the enhanced stability of jcLNA modified oligonucleotides in the blood serum and snake venom phosphodiesterase (SVPDE) digestion, compared to the LNA-modified counterpart (∼100 times) and that of the native (∼1000 times). The reduced catalytic efficiency of jcLNA modified oligos to nucleases (Kcat/Km = 0.0059 μM min−1) is 100 times less than that of LNA counterpart (Kcat/Km = 0.625 μM min−1). This is owing to the fact that the pKa of the departing 3′-oxyanion in jcLNA is 1.4 units higher than the LNA nucleoside.4 The lower Kcat (2.21 μM min−1) of the jcLNA-oligo, compared to that of the LNA counterpart (236.6 μM min−1) is due to the poorer binding affinity (Km) of the former (412.1 μM min−1) compared to the latter (365.5 μM min−1) towards 3′-exonuclease as well as the higher pKa of the 3′-OH of jcLNA, which makes it less labile as a leaving group during 3′-exonuclease mediated degradation than those of the LNA counterparts, thus accounting for the enhanced stability of jcLNA-modified oligos in blood serum.
The silencing efficiency of native, as well as chemically-modified, siRNA molecules has also been found to be determined by other factors such as modification sites, thermodynamic properties, secondary structures, and subsequent accessibility of its target RNA sequence.27
For HIV-1 in particular, designing functional siRNAs that target the viral sequence is challenging because of its extraordinarily high genetic diversity28 and the propensity of the virus to undergo adaptive mutations.29,30 The trans-acting responsive (TAR) element located in the HIV-1 5′-LTR region is a well-described tight stem-loop structure.31 The apical part of TAR acts as a binding site for the HIV-1 trans-activator protein or TAT and some other host cellular factors. This complex triggers the elongation and production of full length HIV transcripts.32 Due to its importance in viral replication, TAR has been an important target for RNAi-based studies.33–35 In this present study we have chosen two targets namely TAR1 and TAR2 for initial validation of target efficacy. The TAR1 sequence has a 16 base overlap with the TAR3 sequence proposed by Yoshinari and co-workers.35 The experimental model described there35 is largely different from the one used in this present study in that they had used TAR constructs instead of a virus model.
The TAR1 sequence of this present study has a 19 base overlap with the TAR4 sequence used by Leonard and co-workers.34 The authors had shown effective inhibition of viral replication in short durations of infection when RNAi is targeted against the TAR region. However, quite uniquely, the authors have given evidence that in long term infections HIV replication is hindered not by direct mutations in the TAR region but by an indirect compensatory mechanism. Another TAR target used in this study, TAR2, has also been successfully utilized in the modulation of HIV-1 replication.33
In the context of therapeutics the bioavailibility of siRNAs, especially in body fluids like serum, would depend much on resistance to RNase A like enzymes and 3′-exonucleases.36,37 Innovative chemistry leading to considerably better 3′-exonuclease resistance of jcLNA over LNA (Fig. 1a and 1b respectively) opens up the avenue for testing jcLNA modified siRNAs in disease models. This is the first exhaustive report of jcLNA modified siRNAs being used in successful inhibition of HIV-1 replication. siRNAs targeting HIV-1 TAR region containing conformationally locked 2′,4′-carbocyclic ribo-Thymidine (jcLNA) modifications2 at various positions have been used and studied for their silencing efficiency as well as stability in serum. jcLNA modified siRNAs used in this study proved to be better in efficiency and presented significantly enhanced stability in human serum.
Results
Relative target potential of sites
To evaluate the potency of the selected sites, namely the TAR1 and TAR2 regions, in our experimental set-up, native siRNAs were synthesised targeting these sites (Table S1, ESI†). The target potential was subsequently analyzed by dose response studies in co-transfection experiments using 100 ng of pNL4-3 and varied doses of the native siRNAs covering a dose range of 0.025 nM to 250 nM in HEK 293T cells. HIV-1 p24 ELISA of culture supernatants 48 h post-transfection revealed dose dependent inhibition of virus formation (Fig. 2).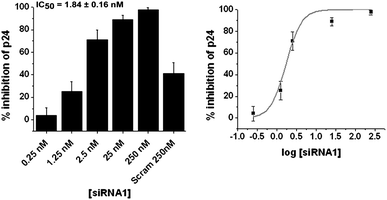 | ||
| Fig. 2 Dose response studies using p24 ELISA for native siRNA1 showing % inhibition over virus control at various doses. The dose titration sigmoidal plot is in the right panel. Results are a cumulative of at least three independent experiments. Error bars represent ±SD from mean. p24 ELISA was carried out with culture supernatants, 48 h post-co-transfection of pNL4-3 and varying doses of siRNA. | ||
To quantitate the target potential of a site, IC50 values were calculated from the dose response data (Fig. 2 and Fig. S1, ESI†). Although the efficacy of chemically modified antisense as anti-HIV agents has been successfully used earlier in low concentration range (0.16–20 nM) targeting the primer binding site (PBS) and dimmer initiation site (DIS) of HIV-1 5′ UTR, no IC50 values were calculated.38 In accordance with this observation and also to negate off-targeting and cytotoxic effects39,40 associated with higher siRNA concentrations, we set a stringent cut-off barrier for IC50 obtained from p24 ELISA of culture supernatant at 10 nM. Since the IC50 value for TAR2 site (targeted by siRNA2) was found to be 22.85 ± 8.96 nM (Fig. S1, ESI†) compared to 1.84 ± 0.16 (Fig. 2) for TAR1 site (targeted by siRNA1), the latter was selected from the two for further validation of results by western blot and RTPCR.
In the intracellular context p24Gag is formed by post-translational proteolytic cleavage of p55Gag polyprotein.41 Hence intracellularly, the effect of RNAi triggering disruption of TAR would be ‘directly’ reflected in the levels of transcripts like Gag mRNA and subsequently in its full length protein amounts (p55Gag). Western blot and RTPCR (qualitative) studies were further performed to study the intracellular protein and mRNA levels of Gag and also to correlate with the ELISA data. The HIV-1 p24 antibody used in this study recognized the p55 as well as p24 fragments of Gag as with other antibodies used in other studies,42,43 the p55Gag recognition being predominant in our experiments. Based on these objectives and conditions densitometry calculations were performed to obtain IC50 values of p55Gag inhibition from western blot (Fig. 3a). The p24Gag profile was observed to be directly proportional to the p55Gag levels (Fig. 3a).
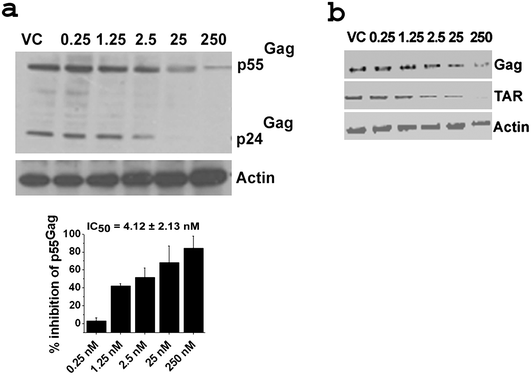 | ||
| Fig. 3 Dose response studies using Western blot and RTPCR for native siRNA1. a: Western blot with corresponding densitometric plot below. For dose titration sigmoidal plots refer to Fig. S2 (ESI†). Results are cumulative of at least two independent experiments. Error bars represent ±SD from mean. b: RTPCR. Concentrations in nM are indicated above the gels. Gels are representative of at least two independent experiments. Western blot was performed with 40 μg of total protein isolated from cells 48 h post co-transfection of pNL4-3 and varying doses of siRNA. The blot was probed with HIV-1 α-p24 and α-Actin antibodies. Band intensities of p55Gag were calculated and normalized with that of actin intensity for each sample using ImageJ software. % inhibition was calculated from the normalized intensity over virus control (VC). RTPCR was performed with 1 μg of RNA isolated from cells 48 h post co-transfection of pNL4-3 and varying doses of siRNA. The PCR was performed with HIV-1 Gag, TAR, Nef and Tat along with human actin specific primers. For western blots and RTPCR of scrambled and mock transfected samples refer to Fig. S3a and S3b (ESI†) respectively. For RTPCR of HIV-1 Nef and Tat refer to Fig. S3c (ESI†). For protocols of western blot and RTPCR see ESI.† | ||
The IC50 value of p55Gag inhibition for siRNA1 from western blot was calculated to be 4.12 ± 2.13 nM and correlated well with that obtained from p24 ELISA (1.84 ± 0.16 nM). RTPCR results displayed dose-dependent down regulation of Gag RNA (represented by amplicons spanning positions 1544 to 1658 of pNL4-3 sequence) (Fig. 3b) as well as TAR levels (represented by amplicons spanning positions 320 to 577 of pNL4-3 sequence and containing the TAR1 site) (Fig. 3b) for siRNA1. Hence, the TAR1 site proved to be a credible target for RNAi mediated inhibition of HIV-1 replication and an ideal one for the introduction of subsequent jcLNA and LNA modifications at various positions of the anti-sense strand (guide strand) of siRNA1 to compare the inhibitory effect of jcLNA vs. LNA. It also appears that upon disruption of the primary target, TAR, a relay in reduction of other viral transcripts like Nef and Tat occurs (Fig. S3c, ESI†).
jcLNA modified siRNA1 targeting TAR1 shows better efficiency (HIV-1 inhibition) than LNA modified siRNA1
jcLNA (Fig. 1a) substituted antisense oligonucleotides (AON) have been shown to be more complimentary RNA-selective by 1–2 °C per substitution3 over LNA (Fig. 1b). Compounded to this the jcLNA modified AONs have been shown to display increased resistance to nucleases (snake venom phosphodiesterase) primarily by hindering the nuclease–phosphate linkage interaction.3 Based on the sequence of the antisense strand of siRNA1, jcLNA-T was substituted for U at different positions (Table S2, ESI†). To examine the activity of the jcLNA modifications parallel LNA representatives were used as controls in a pairwise study (Table S2, ESI†).Subsequently dose response studies were performed with the jcLNA and LNA modified siRNAs as earlier (Fig. 4) and inhibition of virus formation quantified by p24 ELISA of culture supernatants.
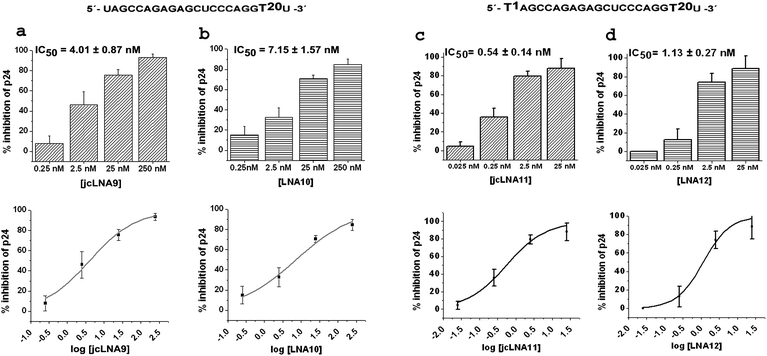 | ||
| Fig. 4 Dose response studies using p24 ELISA for jcLNA and corresponding LNA modified siRNA1 showing % inhibition over virus control at various doses. a: jcLNA9, b: LNA10, c: jcLNA11 and d: LNA12. The sequence for a particular jcLNA/LNA pair is represented above the bar plots with the position(s) of modification highlighted. Respective dose titration sigmoidal plot are placed below each bar plot. Results are cumulative of at least three independent experiments. Error bars represent ±SD from mean. p24 ELISA was carried out with culture supernatants 48 h post-co-transfection of pNL4-3 and varying doses of siRNA. | ||
The dose range remained comparable with that of native siRNA1. Due to the intrinsic differences in silencing efficiency the lowest dose points of jcLNA 3/LNA4, jcLNA5/LNA6 and jcLNA7/LNA8 could not be adjusted to be same (Fig. 4 and S4, ESI†). At the TAR1 site modification at the position 13 only (T13) yielded similar IC50 values for jcLNA3 (IC50 = 8.12 ± 2.06 nM) and LNA4 (IC50 = 11.7 ± 3.85 nM) (Fig. S4a and S4b, ESI†). Introduction of an additional modification at position 1 from the 5′-end (T1 + T13) led to more than a two-fold difference in the IC50 values for jcLNA5 over the corresponding LNA6. The value for jcLNA5 (IC50 = 10.77 ± 1.94 nM) was nearly two-fold lower than the corresponding LNA6 (IC50 = 24.41 ± 1.87 nM) (Fig. S4c and S4d, ESI†). Interestingly, the modification at the extreme 5′-end only (T1) led to comparable IC50 values for both jcLNA and LNA. jcLNA7, having a modification at position 1, displayed marginally higher IC50 than the corresponding LNA8 (Fig. S4e and S4f, ESI†). It thus seems that at the TAR1 site the jcLNA modification at the core region (T13) is not only well tolerated but also increases the efficiency of inhibition than that of the LNA modification when combined with a modification at the 5′-end (T1). The 3′ overhang modifications, crucial in 3′ exonuclease resistance, showed the best inhibition efficiencies. For the 3′-end modification (T20) jcLNA9 shows near native like IC50 value of 4.01 ± 0.87 nM whereas the LNA counterpart LNA10 showed nearly two-fold higher IC50 value of 7.15 ± 1.57 nM (Fig. 4a and 4b respectively). When compounded with position 1 modification (T1 + T20) jcLNA11 showed IC50 value (0.54 ± 0.14 nM, Fig. 4c) which was more than three fold lower than native and two fold lower than LNA12 (1.13 ± 0.27 nM, Fig. 4d). Hence jcLNA11 can be considered as a significant development leading to enhanced efficiency over native.
To correlate the efficacy of the modified siRNAs in down-regulating virus formation with the intracellular transcript levels like Gag, western blot as well as RTPCR was performed, as for native siRNA1. We observed dose-dependent down-regulation of p55Gag as well as its proteolytically cleaved p24Gag fragment levels from the western blot study (Fig. 5 and S5, ESI†), and subsequently the IC50 of p55Gag down regulation was calculated.
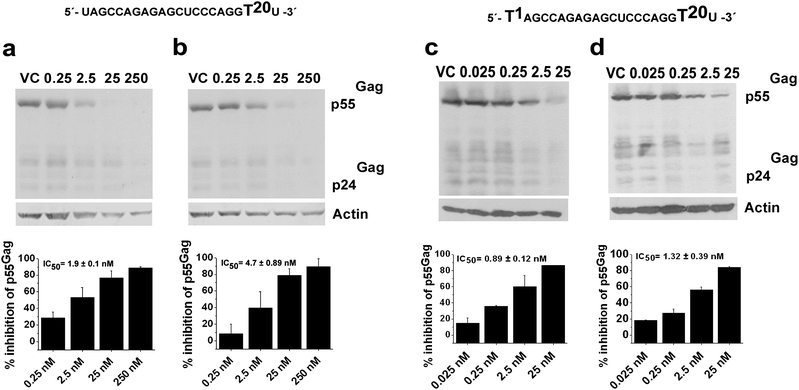 | ||
| Fig. 5 Dose response studies using Western blot for jcLNA and LNA modified siRNA1. a: jcLNA9, b: LNA10, c: jcLNA11 and d: LNA12. The sequence for a particular jcLNA/LNA pair is represented above the blot with the position(s) of modification highlighted. Concentrations in nM are indicated above the gels. For dose titration sigmoidal plots refer to Fig. S6, ESI.† Results are cumulative of at least two independent experiments. Error bars represent ±SD from mean. Western blot was performed with 40 μg of total protein isolated from cells 48 h post co-transfection of pNL4-3 and varying doses of siRNA. The blots were probed with HIV-1 α-p24 and α-Actin antibodies. Band intensities of p55Gag were calculated and normalized with that of actin intensity for each sample using ImageJ software. % inhibition was calculated from the normalized intensity over virus control (VC). For protocols of western blot see ESI.† | ||
The average IC50 values from western blot were largely comparable to those obtained from ELISA (Table 1). Except for jcLNA7/LNA8 and jcLNA11/LNA12 the average IC50 values obtained from western blot for the modifications were marginally lower than those from ELISA study (Table 1). Nevertheless these exceptions were fairly near or within the error range of the respective IC50s from ELISA.
| Site of siRNA Modification | IC50 at TAR1 site (in nM) | t 1/2 in serum (in hours) | ||||
|---|---|---|---|---|---|---|
| jcLNA | LNA | jcLNA | LNA | |||
| ELISA | Western blot | ELISA | Western blot | |||
| T13jcLNA3/LNA4 | 8.12 ± 2.06 | 7.95 ± 1.83 | 11.7 ± 3.85 | 7.89 ± 2.15 | 8.7 | 4.6 |
| T1 + T13jcLNA5/LNA6 | 10.77 ± 1.94 | 4.88 ± 2.64 | 24.41 ± 1.87 | 13.33 ± 7.13 | 5.9 | 6.4 |
| T1jcLNA7/LNA8 | 17.69 ± 1.23 | 22.1 ± 8.23 | 13.66 ± 0.28 | 17.05 ± 0.77 | 4.3 | 3.9 |
| T20jcLNA9/LNA10 | 4.01 ± 0.87 | 1.9 ± 0.1 | 7.15 ± 1.57 | 4.7 ± 0.89 | 11.9 | 6.8 |
| T1 + T20jcLNA11/LNA12 | 0.54 ± 0.14 | 0.89 ± 0.12 | 1.13 ± 0.27 | 1.32 ± 0.39 | 13.6 | 11.9 |
In addition dose dependent down-regulation of Gag RNA was observed in RTPCR studies (Fig. 6 and S7, ESI†). Furthermore, RTPCR was also performed to amplify the HIV-1 TAR region containing the site of interference. Dose dependent reduction of TAR amplicons was observed similar to that of Gag amplicons (Fig. 6 and S7, ESI†).
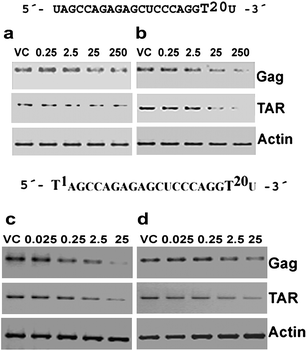 | ||
| Fig. 6 Dose response studies using RTPCR for jcLNA and LNA modified siRNA1. a: jcLNA9, b: LNA10, c: jcLNA11 and d: LNA12. The sequence for a particular jcLNA/LNA pair is represented above the gels with the position(s) of modification highlighted. Concentrations in nM are indicated above the gels. Gels are representative of at least two independent experiments. RTPCR was performed with 1 μg of RNA isolated from cells 48 h post co-transfection of pNL4-3 and varying doses of siRNA. The PCR was performed with HIV-1 Gag and TAR along with human actin specific primers. For protocols of RTPCR see ESI.† | ||
Since, siRNAs have been associated with toxicity39 MTT assay was performed to estimate cell viability after 48 h of transfection. It is evident from p24 ELISA that at the highest dose points more than 80% viral inhibition could be attained for all modified siRNAs and hence, cell viability was measured at these highest doses. Excepting jcLNA11 and jcLNA12, where more than 80% viral inhibition occurred at the highest dose of 25 nM, the cell viability assay for the rest of the siRNAs was carried out at 250 nM. For jcLNA substituted siRNAs at TAR1 site no considerable cytotoxicity was observed compared to LNA substituted siRNAs. More than 70% cell viability was observed for both jcLNA and corresponding LNA candidates (Fig. 7). The viability of the chemically modified siRNAs was similar to native siRNA1 (Fig. 7). This high cell viability with no considerable cytotoxicity is quite interesting because high silencing activity of siRNA is generally accompanied by high cellular toxicity because the observed siRNA toxicity arises from efficient interfering with the endogenous RNAi pathway.9 Accordingly, the similar toxicity of the jcLNA modified siRNAs (7′R/S-Me-carba-LNA modified siRNA), compared to the native siRNA shows that most probably our siRNAs are not interfering with any endogenous pathway. Thus again we show that it is possible to generate carba-LNA modified siRNAs with high down-regulating activity and low toxicity—an encouraging step of a potential therapeutic in the making.
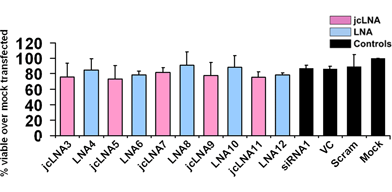 | ||
| Fig. 7 Cell viability assay (MTT assay). jcLNA and LNA modified siRNA1 48 h post transfection 104 cells were subjected to MTT assay in a 96 well plate. Cells having the highest dose of each siRNA (25 nM for jcLNA11/LNA12 and 250 nM for the rest) were used. Error bars represent ±SD from mean. Viability was measured over that of mock transfected cells. Results are cumulative of at least three independent experiments. | ||
jcLNA modification confers exceptional stability in serum in a position dependent manner
Serum stability studies revealed unprecedented stability of jcLNA modified ds siRNAs. Out of five types of modifications two jcLNA modified siRNAs had considerably higher t1/2 values than LNA counterparts and for the other three, jcLNA and LNA showed perfectly similar t1/2 values. All the modifications, jcLNA or LNA, displayed considerably better t1/2 values than the native. Native double strand (ds) siRNA1 displayed stability until 90 min of incubation in human serum, degradation being apparent from 4 h and practically degrades fully at 12 h (Fig. S8a, ESI†). The calculated t1/2 value for native ds siRNA1 is 3.0 h (Fig. S8a, ESI†).For jcLNA3, having modification at position 13 (T13), prominent degradation is apparent from 6 h but the integrity of its predominant ds form is retained until 12 h (Fig. S8b, ESI†); degradation of ds form being complete at 24 h. On the other hand for the corresponding LNA4 degradation of ds form is visible as early as 4 h ultimately leading to near total annihilation of the ds form at 12 h (Fig. S8c, ESI†). For this position 13 modification the t1/2 value for jcLNA3 is 8.7 h in contrast to 4.6 h of LNA4. Another considerable modification in terms of t1/2 value is position 20 (T20), implicated in enhanced resistance towards exonucleases. jcLNA9, with modification at position 20, shows robust retention of its ds form till 12 h (Fig. 8a) whereas LNA10 does so till 6 h (Fig. 8b). In accordance with this observation the t1/2 value of jcLNA9 is 11.9 h and considerably better than 6.8 h for counterpart LNA10 (Fig. 8a and 8b). Conjunction of position 1 modification with position 20 (T1 + T20) led to further improvement of t1/2 value for jcLNA11 as well as for LNA12. Although the degradation profile appears nearly similar LNA12 (t1/2 = 11.9 h, Fig. 8d) shows higher degradation at 24 h compared to jcLNA11 (t1/2 = 13.6 h, Fig. 8c). Both jcLNA5 and LNA6, having double modifications at position 1 and 13 (T1 + T13), displayed considerable stability for 12 h (Fig. S8d and S8e, ESI†) displaying t1/2 values of nearly 6 h each. Similarly, for both jcLNA7 and LNA8, having modification at position 1 (T1), considerable integrity of the ds form is apparent for 6 h (Fig. S8f and S8g, ESI†) and the t1/2 values for both are practically same, being ∼4 h (Fig. S8f and S8g, ESI†).
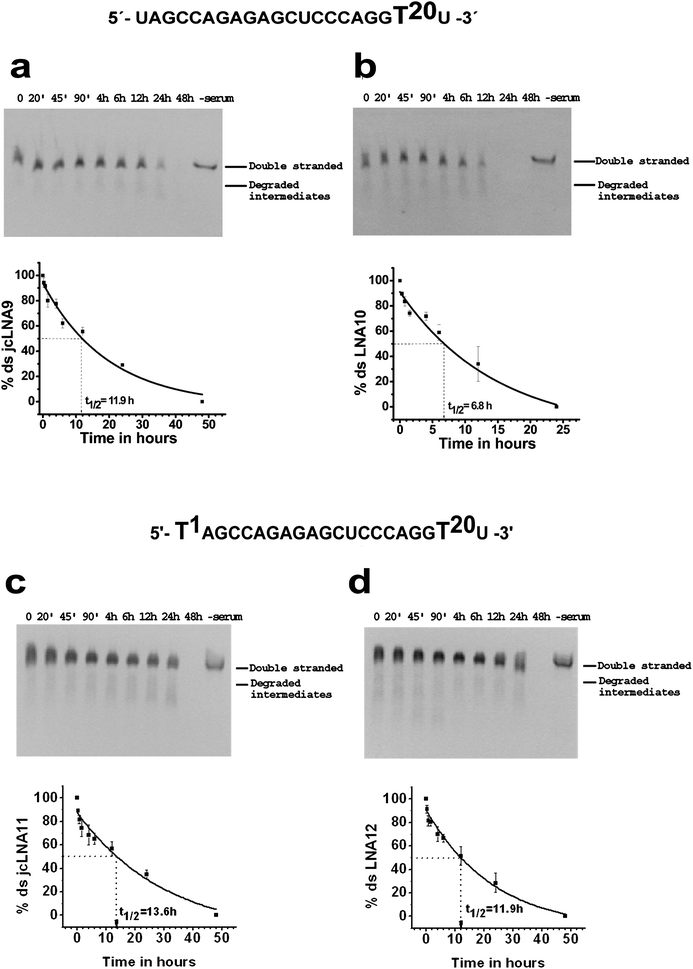 | ||
| Fig. 8 Serum stability of chemically modified double stranded siRNAs targeting TAR1 at different time points. a: jcLNA9, b: LNA10, c: jcLNA11 and d: LNA12. The sequence for a particular jcLNA/LNA pair is represented above the gels with the position(s) of modification highlighted. Respective best fit exponential decay curve of the mean % double stranded form showing the mean t1/2 value is represented below the gels. Gels and curves are representative of at least three experimental repeats. Error bars represent ±SD from mean. | ||
Discussion
In the quest for established targets in HIV-1 to test the efficacy of jcLNA and LNA modified siRNAs we had initially selected two previously described target sites of the HIV-1 TAR region.33,34 The effect of RNAi, independent of the interference target, resulting in the inhibition of virus formation has been quantitated by p24Gag content of culture supernatant by ELISA.44,45 In the initial screen involving the two native siRNAs namely siRNA1 and siRNA2 in our transient co-transfection and single cycle replication model we observed an average IC50 of virus inhibition varying from 1.84 nM to 22.85 nM respectively. Subsequently, the low IC50 values associated with siRNA1 made it a suitable candidate for introduction of chemically modified nucleotides namely jcLNA and LNA. With an objective to study the effect of interference at TAR on the intracellular HIV-1 gene expression we calculated the IC50 of p55Gag (pre-proteolysis precursor of p24Gag) inhibition from dose response studies. In agreement with ELISA (of culture supernatant) results, dose dependent down regulation of intracellular p55Gag was observed both at the protein, as well as at the RNA level, for siRNA1; the proteolysed intracellular p24Gag fragment was found to be proportional to the p55 fragment. The IC50 of virus inhibition (obtained from ELISA) was generally comparable with that of intracellular p55Gag inhibition. Dose dependent reduction of target amplicons, namely TAR, was also observed by RTPCR for siRNA1 reflecting its target affinity. Jacque and co-workers33 had indicated in their study that the viral genomic RNA is susceptible to specific degradation via RNAi and subsequently the provirus integration in the human genome is thwarted. Yet another study46 had strongly indicated the contrary, primarily implicating the tightly packaged viral core encapsulating the RNA genome thereby rendering the viral RNA inaccessible to RNAi machinery. In our study, the nascent mRNA transcribed from pNL4-3, obviously devoid of any viral packaging, may be susceptible to RNAi and hence a global down-regulation of HIV genes may occur as reflected by decreasing levels of intracellular p55Gag for siRNA1. In this context we have observed dose dependent reduction of Nef and Tat amplicons in RTPCR experiments when TAR1 had been targeted by siRNA1. This strongly indicates a global down-regulation of HIV genes in our current study when RNAi is directed at the well characterized TAR1 target.The quintessential requirement of thermodynamic stability compounded with resistance to nucleases, while not compromising the specificity, has triggered research for chemically modified nucleotides substituting the normal ones in siRNA. These chemically-modified siRNAs have also one other important goal, to improve its delivery to the cell. LNA [2′-O,4′-C-methylene-β-D-ribofuranosyl] monomers have been successfully used in gene down-regulation whose efficacy is enhanced by high-affinity hybridization to ssDNA as well as ssRNA,47–51 serum stability, efficient RISC loading23 and nuclease resistance.52 Based on these established properties, LNA modified siRNAs were used in this study for comparison of efficiency of jcLNA substituted siRNA1 in the inhibition of HIV-1 replication. Our results primarily indicate that in our experimental model the inhibition efficiency of jcLNA as well as LNA modified siRNAs was generally less than the native counterpart as reflected by the IC50 values. However, jcLNA T1 + T20 double modification of the antisense strand (first base from 5′-end and second base in 3′-end overhang) surpassed the inhibition efficiency of the native by over three fold and more than two fold over the LNA counterpart (Table 1). Also, solitary T20 jcLNA modification displayed near native-like efficiency in terms of IC50 values; jcLNA being better than its LNA counterpart by nearly two fold (Table 1). In a recent study9 antisense strands modified exclusively at the 3′-overhangs with LNA, EA (2′-aminoethyl), ALN (α-L-LNA) and ANA (altritol nucleic acid) moieties were found to be present in almost 50% of the highly efficient siRNAs, although jcLNA modification at this position was not used in the study. Our study comprehensively adds jcLNA modified 3′-antisense overhang siRNAs to this efficient class of siRNAs. Our study further revealed that jcLNA modification introduced outside the seeding region (2–8 of the antisense strand53); at position 13 of the antisense strand is equally tolerated vis-à-vis the LNA counterpart. In yet another observation we found that for both jcLNA and LNA inhibition efficiency was considerably jeopardized when the modification was introduced at position 1 of the antisense strand (first base from the 5′-end) (Table 1). It had been previously noted23 that LNA incorporation at the 5′-antisense end is associated with loss of activity and that such a negative phenomenon could be balanced by similar modification at the 5′-sense end. Clearly our results, too, hint at such a loss of activity with both jcLNA and LNA substitutions at this position. However, conjunction of position 13 and position 1 modifications (double substitution) makes the IC50 of jcLNA lower than that of the LNA counterpart by over two fold (Table 1). In other terms, for jcLNA, there is a considerable rescue in loss of silencing efficiency stemming from position 1 modification when the same is compounded with position 13 modification. Quite interestingly, overhang T20 modification in conjunction with T1 not only rescues silencing activity relative to T1 but also betters that of the native for both jcLNA and LNA. It is worth mentioning at this point is that such a combinatorial effect would depend not only on the local chemistry of the interaction between the modified nucleotides and the target but also on the geometry/folding of the target RNA itself; hence further validation with different targets can address the universality of this combination. Nonetheless our study indicates that at TAR1 site jcLNA modified siRNAs are either largely equal in efficiency to LNA or considerably better in a position dependent manner and at least in one case displays HIV-1 inhibition efficiency better than native.
The IC50 values in our study are based on a 48 h window of experimentation post-transfection. For actively dividing cells the dilution of intracellular native siRNAs has been shown to affect silencing, which generally lasts for a week36,54,55 (or up to 3 weeks for non-dividing cells36). Hence, as an obvious corollary in our case the IC50 values might have been higher if the experimentation had been carried out for a week. Although the intracellular activity of native siRNAs may extend up to a week in vitro, their stability and integrity in serum is in the order of hours.23 At this point the significance of chemically modified siRNAs comes into play and results have shown considerable enhancement of integrity of modified siRNAs9,23 or AONs2,3 in serum over the native ones. In our present study the ds form of all the modifications, as expected, yielded higher t1/2 values in serum than native siRNA1 whose value was calculated to be 3.0 h. Direct correlation of stability with inhibition efficiency can be observed for jcLNA substituted siRNAs. For the T1 + T20 and T20 overhang modifications jcLNA displayed unprecedented stability in serum presenting t1/2 values surpassing that of LNA as well as that of the native (Table 1). Notably T1 modification alone, for both jcLNA as well as LNA, yields the worst t1/2 values (4.3 h and 3.9 h respectively) amongst all the modifications. When T20 is combined with this the t1/2 shoots up to 13.6 h for jcLNA and 11.9 h for LNA (Table 1). Hence, T20 modification at the 3′-antisense overhang plays a crucial role in siRNA protection. This is additionally proven by the fact that T20 modification alone also confers tremendous stability at least for jcLNA with a t1/2 of 11.9 h compared to 6.8 h for LNA (Table 1). Furthermore T1 + T13 double modification does not emphatically rescue stability for both jcLNA and LNA relative to T1; hence substantiating strongly the necessity of 3′-antisense end protection. As a minor deviation we observed a fair display of stability of jcLNA T13 modification (t1/2 of 8.7 h, Table 1) compared to that of T1 modification, both having unprotected 3′-antisense end. In the therapeutic context the integrity of ds siRNA in body fluids like serum would be of paramount importance for efficient post-delivery inhibition of target sites in targeted cells. RNase A like enzymes have been implicated in the degradation of ds siRNAs in serum37,56 and in humans pancreatic ribonuclease (hpRNase) has been proposed as a likely candidate totalling 70–80% of total ribonuclease activity in serum.57–59 In this context Haupenthal and co-workers had shown that 3′-overhangs render siRNAs susceptible to RNaseA like enzymes.37 The considerably better serum stability as well as efficiency of 3′-overhang jcLNA modified ds siRNAs over LNA makes them distinguished modifications. The report of conserved siRNase in C. elegans60 makes the role of cellular endo and exonucleases equally crucial even if a siRNA makes its way to a target cell. Work is currently on in our laboratory to analyse the stability of modified ds siRNAs in cellular extracts also.
Moreover, cells transfected with jcLNA modified siRNAs, in our experimental model, shows at least 70% cell viability (even at 250 nM concentration) and comparable with that observed with LNA substituents. This addresses the problem of cytotoxicity associated with siRNAs39,61,62 which may be a potential pitfall in the use of RNAi as a therapeutic tool. Off-targeting side-effect of siRNAs is yet another undesirable phenomenon. A recent work63 had reported position specific reduction in off-targeting by UNA modified siRNAs. The authors had also shown that position 3 modification of antisense strand by jcLNA, CENA, HNA and EA potently reduces off-targeting. The off-targeting potential, if any, of jcLNA modified siRNAs used in this present study would be a pertinent issue and work is currently in progress to address the same.
Conclusion
This is the first study showing the exploitation of jcLNA modified siRNAs against HIV-1 targeting the TAR region based on single cycle replication model. Through this successful study we have exhaustively brought forth the immense therapeutic potential of jcLNA modified siRNAs in a single target intervention approach. We herein show that amongst all the modified siRNAs so far used to down-regulate the RNA targeting HIV-1 TAR region, the best IC50 value was obtained in the modified siRNAs with jcLNA both at position 1 and 20 of the guide strand (T1 + T20), as in jcLNA11 which showed IC50 value of 0.54 ± 0.14 nM (Fig. 4c). The IC50 of this doubly-modified siRNA was more than ca 3-fold lower than that of the native (1.84 ± 0.16 nM) and two-fold lower than that of LNA modified counterpart, i.e. LNA12 (1.13 ± 0.27 nM, Fig. 4d). Hence the strategy to modify the native siRNAs with the jcLNA can be considered as a significant development, leading to both enhanced siRNA efficiency and serum stability over that of the native. In some other cases the silencing efficiency with jcLNA modified siRNAs was not native-like but it was effectively compensated by the enhanced stability in serum. Most interestingly, the 3′-end modified siRNAs yielded silencing efficiency comparable to that of the native; clearly owing to higher stability. The hallmark of HIV infection is the ability of the virus to mutate and thus resist target directed therapeutics. Combinatorial RNAi against various RNA targets of the HIV has been projected as a future therapeutic procedure.64,65 Hence, a multi-target intervention using smart positional jcLNA modification could radically enhance the potency of siRNA based treatment.Implication
We have synthesised various kinds of jcLNA (carba-LNA) modified siRNAs which we have targeted to HIV-1 TAR region using HIV-1 single cycle replication model. Our results demonstrate that jcLNA modifications in the siRNA have the potential to inhibit virus replication with maximum efficiency and target specificity. In addition, jcLNA modified siRNAs display robust stability in biological fluids and negligible cytotoxicity. The real potential of carba-LNAs is in its chemical construction in that, because of its pendant alkane bridge 4′–CH2–CH(Me)–2′ fused covalently to the C-2′ and C-4′ of the ribose unit, it allows substitution of many types of hydrophobic or hydrophilic groups on the alkane chain with defined stereochemistry, thereby modulating various substituent and stereochemistry-dependent pharmacokinetic properties expected from a potential RNA-directed therapeutic. To achieve this goal, we have already introduced2–4 various chemical methodologies for specific functionalization on the pendant 4′–CH2–CH2–2′ chain, and have also preliminarily elaborated their effects as modified oligonucleotides on the serum stability as well as on thermodynamic stability of their heteroduplexes with RNA. Further work is in progress to examine how we can improve the siRNAs with these modifications for targeting RNA. Hence, jcLNA modified siRNAs described in this work not only open a promising therapeutic possibility in the HIV-1 treatment, as shown in this study, but also against other infectious or genetic diseases.Experimental section
Synthesis, Deprotection and purification of Oligonucleotides
All oligonucleotides were synthesized using an automated DNA/RNA synthesizer by Applied Biosystems, model 394. The stepwise coupling yields of the modified phosphoramidites i.e. LNA-T (Link Technologies) and jcLNA-T2 were 96% and 98% respectively. 5-(3,5-bis(trifluoromethyl)phenyl)-1H-tetrazole (Activator 42) as the activating reagent with 10 min coupling time for modified phosphoramidites, followed by deprotection of all base-labile protecting groups with 33% methanolic ammonia at 55 °C to give oligonucleotides. All oligonucleotides were purified by (20% polyacrylamide/7 M urea) PAGE, extracted with 0.3M NaOAc, desalted with C18-reverse phase cartridges (Waters) and their purity (greater than 95%) was confirmed by PAGE. Concentrations were determined by diluting the stock solutions in DEPC-treated water followed by analysis using a UV spectrophotometer (Shimadzu UV2550).Duplex formation
RNA duplexes were prepared by the annealing of complementary oligonucleotides in thin-walled PCR tubes containing same concentrations of sense and antisense (equimolar) of each in DEPC treated water and annealing buffer (5X; 50 mM Tris; pH 7.5–8.0; 100 mM NaCl in DEPC-treated water). Annealing of RNA duplexes was performed in a thermal cycler by incubating the solution for 1 min at 90 °C followed by slowly cooling to room temperature over a period of about 60 min. After annealing, the RNA duplexes were stored at −20 °C. List of siRNAs synthesised are given in Table S1 and Table S2, ESI.†Cell culture and transfection
HEK293T cells were cultured in DMEM (Sigma) supplemented with 10% bovine serum (PAN Biotech) and 1% pencillin–streptomycin solution (Sigma). Cells were grown at 37 °C in presence of 5% CO2. 24 h prior to transfection 105 cells/well were seeded in a 24 well plate or 3 × 105 cells/well were seeded in a 6 well plate. Subsequently, siRNAs and 100 ng pNL4-3/well were co-transfected in 24 well plate using Lipofectamine 2000 (Invitrogen) as per the manufacturer's protocol. For transfection in 6 well plates the calculations were stepped up three times per well.p24 ELISA of culture supernatant
p24 ELISA of culture supernatant was performed using Perkin Elmer Alliance HIV-1 p24 antigen ELISA kit as per the manufacturer's recommendation. The mean percentage inhibitions along with standard deviation were calculated with respect to the OD obtained from the virus control (VC) samples in which pNL4-3 was transfected only from at least three independent experiments. The mean percentage inhibitions plotted against the logarithm of respective concentrations were used to calculate IC50 value using the sigmoidal curve fitting dose response option of Origin® 6.1 software (http://www.originlab.com/) based on the logistical function y = [(A1 − A2)/(1 + x/x0)p] + A2 using the default settings of the software. The IC50 value and the error [SQRT(covii*(ChiSqr/DOF))] as calculated by the software are represented.MTT assay
48 h post transfection the supernatant was removed and fresh media was added to each well of a 24 well plate. MTT was added to a final concentration of 0.1 mg ml−1. Subsequently, the cells were incubated for 2.5 h at 37 °C in a CO2 incubator. Following this incubation the reactions were stopped by addition of stop solution (99% Isopropanol and 1% HCl) and OD was measured at 595 nm. Percentage viability was calculated over that of the mock transfected sample and results are a cumulative of at least three independent experiments.Stability assay of siRNAs in human serum
1.5 μl of native as well as jcLNA and LNA modified siRNAs from 100 μM stock were added to 10 μl of 100% human serum isolated from healthy B+ donor in a final volume of 11.5 μl. Samples were incubated for required time points after which 3 μl aliquots were withdrawn. The aliquots were immediately suspended in 9 μl of 1X TBE gel loading buffer (18 mM EDTA, 5% glycerol 0.025% bromophenol blue) snap frozen in liquid nitrogen and stored at −80 °C. The samples were subjected to 20% non-denaturing TBE-PAGE. Following the run gels were stained with ethidium bromide and visualized and documented in a BIORAD Geldoc. Band intensities of the double strand (ds) form were obtained using ImageJ software66,67 and percentage of ds form remaining at each time point was calculated over that at zero time point. The mean percentages of ds form remaining for a particular time point along with standard deviation was calculated from at least three independent experiments. The plot of mean % ds form versus time was fitted on to the exponential decay curve following the equation y = y0 + Ae−x/t using Origin® 6.1 software. The t1/2 value was obtained by extrapolating the 50% ds form to the best fit curve.Abbreviations
| LNA | locked nucleic acids |
| jcLNA | carbaLNA |
| TAR | trans activation response |
Note added after first publication
This article replaces the version published on 27 January 2011, which contained errors in Fig. 7.Acknowledgements
We thank Uppsala University, Sweden for valuable scientific support through Prof. Jyoti Chattopadhyaya. We thank Department of Science and Technology, Government of India, for their generous support of this project (Grant no. VII-PRDSF/109/05-06/TDT dated 31.03.2006). We also thank TCG Life Sciences, Kolkata for funding the collaboration.References
- K. T. Gagnon, H. M. Pendergraff, G. F. Deleavey, E. E. Swayze, P. Potier, J. Randolph, E. B. Roesch, J. Chattopadhyaya, M. J. Damha, C. F. Bennett, C. Montaillier, M. Lemaitre and D. R. Corey, Biochemistry, 2010, 49, 10166–10178 CrossRef CAS.
- P. Srivastava, J. Barman, W. Pathmasiri, O. Plashkevych, M. Wenska and J. Chattopadhyaya, J. Am. Chem. Soc., 2007, 129, 8362–8379 CrossRef.
- J. Xu, Y. Liu, C. Dupouy and J. Chattopadhyaya, J. Org. Chem., 2009, 74, 6534–6554 CrossRef CAS.
- C. Zhou and J. Chattopadhyaya, J. Org. Chem., 2010, 75, 2341–2349 CrossRef CAS.
- C. Zhou, Y. Liu, M. Andaloussi, N. Badgujar, O. Plashkevych and J. Chattopadhyaya, J. Org. Chem., 2009, 74, 118–134 CrossRef CAS.
- A. Nykanen, B. Haley and P. D. Zamore, Cell, 2001, 107, 309–321 CrossRef CAS.
- H. Xia, Q. Mao, H. L. Paulson and B. L. Davidson, Nat. Biotechnol., 2002, 20, 1006–1010 CrossRef CAS.
- D. Cejka, D. Losert and V. Wacheck, Clin. Sci., 2006, 110, 47–58 Search PubMed.
- J. B. Bramsen, M. B. Laursen, A. F. Nielsen, T. B. Hansen, C. Bus, N. Langkjaer, B. R. Babu, T. Hojland, M. Abramov, A. Van Aerschot, D. Odadzic, R. Smicius, J. Haas, C. Andree, J. Barman, M. Wenska, P. Srivastava, C. Zhou, D. Honcharenko, S. Hess, E. Muller, G. V. Bobkov, S. N. Mikhailov, E. Fava, T. F. Meyer, J. Chattopadhyaya, M. Zerial, J. W. Engels, P. Herdewijn, J. Wengel and J. Kjems, Nucleic Acids Res., 2009, 37, 2867–2881 CrossRef CAS.
- D. Bumcrot, M. Manoharan, V. Koteliansky and D. W. Sah, Nat. Chem. Biol., 2006, 2, 711–719 CrossRef CAS.
- J. K. Watts, G. F. Deleavey and M. J. Damha, Drug Discovery Today, 2008, 13, 842–855 CrossRef CAS.
- M. Egli, G. Minasov, V. Tereshko, P. S. Pallan, M. Teplova, G. B. Inamati, E. A. Lesnik, S. R. Owens, B. S. Ross, T. P. Prakash and M. Manoharan, Biochemistry, 2005, 44, 9045–9057 CrossRef CAS.
- M. A. Maier, J. M. Leeds, G. Balow, R. H. Springer, R. Bharadwaj and M. Manoharan, Biochemistry, 2002, 41, 1323–1327 CrossRef CAS.
- M. Manoharan, Biochim. Biophys. Acta, Gene Struct. Expression, 1999, 1489, 117–130 CrossRef CAS.
- M. Teplova, S. T. Wallace, V. Tereshko, G. Minasov, A. M. Symons, P. D. Cook, M. Manoharan and M. Egli, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14240–14245 CrossRef CAS.
- N. Albaek, M. Petersen and P. Nielsen, J. Org. Chem., 2006, 71, 7731–7740 CrossRef.
- G. Enderlin and P. Nielsen, J. Org. Chem., 2008, 73, 6891–6894 CrossRef CAS.
- D. Honcharenko, J. Barman, O. P. Varghese and J. Chattopadhyaya, Biochemistry, 2007, 46, 5635–5646 CrossRef CAS.
- M. Kuwahara, S. Obika, H. Takeshima, Y. Hagiwara, J. Nagashima, H. Ozaki, H. Sawai and T. Imanishi, Bioorg. Med. Chem. Lett., 2009, 19, 2941–2943 CrossRef CAS.
- K. Morita, C. Hasegawa, M. Kaneko, S. Tsutsumi, J. Sone, T. Ishikawa, T. Imanishi and M. Koizumi, Bioorg. Med. Chem. Lett., 2002, 12, 73–76 CrossRef CAS.
- S. M. Rahman, S. Seki, S. Obika, H. Yoshikawa, K. Miyashita and T. Imanishi, J. Am. Chem. Soc., 2008, 130, 4886–4896 CrossRef CAS.
- P. P. Seth, A. Siwkowski, C. R. Allerson, G. Vasquez, S. Lee, T. P. Prakash, G. Kinberger, M. T. Migawa, H. Gaus, B. Bhat and E. E. Swayze, Nucleic Acids Symp. Ser., 2008, 52, 553–554 Search PubMed.
- J. Elmen, H. Thonberg, K. Ljungberg, M. Frieden, M. Westergaard, Y. Xu, B. Wahren, Z. Liang, H. Orum, T. Koch and C. Wahlestedt, Nucleic Acids Res., 2005, 33, 439–447 CrossRef CAS.
- O. R. Mook, F. Baas, M. B. de Wissel and K. Fluiter, Mol. Cancer Ther., 2007, 6, 833–843 CrossRef CAS.
- C. Wahlestedt, P. Salmi, L. Good, J. Kela, T. Johnsson, T. Hokfelt, C. Broberger, F. Porreca, J. Lai, K. Ren, M. Ossipov, A. Koshkin, N. Jakobsen, J. Skouv, H. Oerum, M. H. Jacobsen and J. Wengel, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5633–5638 CrossRef CAS.
- C. Thibaudeau, P. Acharya, & J. Chattopadhyaya, Stereoelectronic Effects in Nucleosides and nucleotides and their structural implications, Uppsala University Press, Uppsala, 1999 Search PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
- Y. Naito, K. Nohtomi, T. Onogi, R. Uenishi, K. Ui-Tei, K. Saigo and Y. Takebe, Retrovirology, 2007, 4, 80 CrossRef.
- M. J. Dapp, C. L. Clouser, S. Patterson and L. M. Mansky, J. Virol., 2009, 83, 11950–11958 CrossRef CAS.
- E. Domingo, E. Martinez-Salas, F. Sobrino, J. C. de la Torre, A. Portela, J. Ortin, C. Lopez-Galindez, P. Perez-Brena, N. Villanueva and R. Najera, et al. , Gene, 1985, 40, 1–8 CrossRef CAS.
- H. Van Melckebeke, M. Devany, C. Di Primo, F. Beaurain, J. J. Toulme, D. L. Bryce and J. Boisbouvier, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9210–9215 CrossRef CAS.
- D. Harrich, C. Ulich and R. B. Gaynor, J. Virol., 1996, 70, 4017–4027 CAS.
- J. M. Jacque, K. Triques and M. Stevenson, Nature, 2002, 418, 435–438 CrossRef CAS.
- J. N. Leonard, P. S. Shah, J. C. Burnett and D. V. Schaffer, Cell Host Microbe, 2008, 4, 484–494 CrossRef CAS.
- K. Yoshinari, M. Miyagishi and K. Taira, Nucleic Acids Res., 2004, 32, 691–699 CrossRef CAS.
- D. W. Bartlett and M. E. Davis, Nucleic Acids Res., 2006, 34, 322–333 CrossRef CAS.
- J. Haupenthal, C. Baehr, S. Kiermayer, S. Zeuzem and A. Piiper, Biochem. Pharmacol., 2006, 71, 702–710 CrossRef CAS.
- M. R. Jakobsen, J. Haasnoot, J. Wengel, B. Berkhout and J. Kjems, Retrovirology, 2007, 4, 29 CrossRef.
- Y. Fedorov, E. M. Anderson, A. Birmingham, A. Reynolds, J. Karpilow, K. Robinson, D. Leake, W. S. Marshall and A. Khvorova, RNA, 2006, 12, 1188–1196 CrossRef CAS.
- A. L. Jackson, S. R. Bartz, J. Schelter, S. V. Kobayashi, J. Burchard, M. Mao, B. Li, G. Cavet and P. S. Linsley, Nat. Biotechnol., 2003, 21, 635–637 CrossRef CAS.
- R. W. Humphrey, A. Ohagen, D. A. Davis, T. Fukazawa, H. Hayashi, S. Hoglund, H. Mitsuya and R. Yarchoan, Antimicrob. Agents Chemother., 1997, 41, 1017–1023 CAS.
- S. Abdurahman, S. Hoglund, L. Goobar-Larsson and A. Vahlne, J. Gen. Virol., 2004, 85, 2903–2913 Search PubMed.
- H. A. Baird, A. J. Marozsan, M. M. Lederman, A. Landay, D. Mildvan, D. R. Kuritzkes, H. A. Kessler and E. J. Arts, AIDS Res. Ther., 2005, 2, 2 Search PubMed.
- D. Boden, O. Pusch, R. Silbermann, F. Lee, L. Tucker and B. Ramratnam, Nucleic Acids Res., 2004, 32, 1154–1158 CrossRef CAS.
- K. V. Morris, C. H. Chung, W. Witke and D. J. Looney, RNA Biol., 2005, 2, 17–20 Search PubMed.
- E. M. Westerhout, O. ter Brake and B. Berkhout, Retrovirology, 2006, 3, 57 CrossRef.
- K. Bondensgaard, M. Petersen, S. K. Singh, V. K. Rajwanshi, R. Kumar, J. Wengel and J. P. Jacobsen, Chem.–Eur. J., 2000, 6, 2687–2695 CrossRef CAS.
- D. A. Braasch, Y. Liu and D. R. Corey, Nucleic Acids Res., 2002, 30, 5160–5167 CrossRef CAS.
- A. N. Elayadi, D. A. Braasch and D. R. Corey, Biochemistry, 2002, 41, 9973–9981 CrossRef CAS.
- A. A. Koshkin, J. Fensholdt, H. M. Pfundheller and C. Lomholt, J. Org. Chem., 2001, 66, 8504–8512 CrossRef CAS.
- L. Kvaerno, R. Kumar, B. M. Dahl, C. E. Olsen and J. Wengel, J. Org. Chem., 2000, 65, 5167–5176 CrossRef CAS.
- K. Nagahama, R. N. Veedu and J. Wengel, Bioorg. Med. Chem. Lett., 2009, 19, 2707–2709 CrossRef CAS.
- Y. Wang, G. Sheng, S. Juranek, T. Tuschl and D. J. Patel, Nature, 2008, 456, 209–213 CrossRef CAS.
- C. D. Novina, M. F. Murray, D. M. Dykxhoorn, P. J. Beresford, J. Riess, S. K. Lee, R. G. Collman, J. Lieberman, P. Shankar and P. A. Sharp, Nat. Med., 2002, 8, 681–686 CAS.
- T. Tuschl, Nat. Biotechnol., 2002, 20, 446–448 CrossRef CAS.
- J. J. Turner, S. W. Jones, S. A. Moschos, M. A. Lindsay and M. J. Gait, Mol. BioSyst., 2007, 3, 43–50 RSC.
- M. Libonati and S. Sorrentino, Methods Enzymol., 2001, 341, 234–248 CAS.
- S. Sorrentino, M. Naddeo, A. Russo and G. D'Alessio, Biochemistry, 2003, 42, 10182–10190 CrossRef CAS.
- J. L. Weickmann and D. G. Glitz, J. Biol. Chem., 1982, 257, 8705–8710 CAS.
- S. Kennedy, D. Wang and G. Ruvkun, Nature, 2004, 427, 645–649 CrossRef CAS.
- M. Sioud, Trends Mol. Med., 2006, 12, 167–176 CrossRef CAS.
- C. A. Sledz, M. Holko, M. J. de Veer, R. H. Silverman and B. R. Williams, Nat. Cell Biol., 2003, 5, 834–839 CrossRef CAS.
- J. B. Bramsen, M. M. Pakula, T. B. Hansen, C. Bus, N. Langkjaer, D. Odadzic, R. Smicius, S. L. Wengel, J. Chattopadhyaya, J. W. Engels, P. Herdewijn, J. Wengel and J. Kjems, Nucleic Acids Res., 2010, 38, 5761–5773 CrossRef CAS.
- D. Grimm and M. A. Kay, Mol. Ther., 2007, 15, 878–888 CAS.
- K. J. von Eije, O. ter Brake and B. Berkhout, J. Virol., 2008, 82, 2895–2903 CrossRef CAS.
- Z. Pavicevic, C. C. Leslie and K. U. Malik, J. Lipid Res., 2008, 49, 724–737 CAS.
- C. M. Trivedi, M. M. Lu, Q. Wang and J. A. Epstein, J. Biol. Chem., 2008, 283, 26484–26489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0md00225a |
| This journal is © The Royal Society of Chemistry 2011 |