Sulfonyl-hydrazones of cyclic imides derivatives as potent inhibitors of the Mycobacterium tuberculosisprotein tyrosine phosphatase B (PtpB)†
Kely
Navakoski de Oliveira
a,
Louise Domeneghini
Chiaradia
a,
Priscila Graziela
Alves Martins
b,
Alessandra
Mascarello
a,
Marlon Norberto
Sechini Cordeiro
a,
Rafael Victorio
Carvalho Guido
c,
Adriano Defini
Andricopulo
c,
Rosendo Augusto
Yunes
a,
Ricardo José
Nunes
a,
Javier
Vernal
b and
Hernán
Terenzi
*b
aLaboratório Estrutura e Atividade, Departamento de Química, LEAT-CFM-UFSC, Universidade Federal de Santa Catarina, Campus Trindade, 88040-900, Florianópolis, SC, Brasil
bCentro de Biologia Molecular Estrutural, Departamento de Bioquímica, CEBIME-CCB-UFSC, Universidade Federal de Santa Catarina, Campus Trindade, 88040-900, Florianópolis, SC, Brasil. E-mail: hterenzi@ccb.ufsc.br; Fax: +55 48 3721 6973; Tel: +55 48 3721 6426
cLaboratório de Química Medicinal e Computacional, Centro de Biotecnologia Molecular Estrutural, Instituto de Física de São Carlos, Universidade de São Paulo, Av. Trabalhador São-carlense 400, 13560-970, São Carlos, SP, Brazil
First published on 17th March 2011
Abstract
Searching lead compounds for new antituberculosis drugs, the activity of synthetic sulfonamides and sulfonyl-hydrazones were assayed for their potential inhibitory activity towards a protein tyrosine phosphatase from Mycobacterium tuberculosis – PtpB. Four sulfonyl-hydrazones N-phenylmaleimide derivatives were active (compounds 14, 15, 19 and 21), and the inhibition of PtpB was found to be competitive with respect to the substrate p-nitrophenyl phosphate. Structure-based molecular docking simulations were performed and indicated that the new inhibitor candidates showed similar binding modes, filling the hydrophobic pocket of the protein by the establishment of van der Waals contacts, thereby contributing significantly to the complex stability.
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (Mtb). TB is responsible for the death of two million people worldwide each year, and for the appearance of around 10 million new cases annually.1 Currently, there are drugs with significantly different chemical structures that affect different essential metabolic pathways of Mtb.2 Moreover, multi-resistant strains of Mtb have slowly emerged, providing a long and costly treatment to patients.3–6 For this reason, there is an urgent need for anti-TB drugs with enhanced activity against multidrug resistant (MDR) strains, reduced toxicity and of reduced time for effective therapy.
As a therapeutic strategy, the inhibition of proteintyrosine phosphatases (PTPs), as PtpB and PtpA exoenzymes, is highly promising (for reviews, see ref. 7–9). These enzymes are secreted into the host cell by Mtb and attenuate host immune defenses by interfering in the host signaling pathways.10,11 According to Singh and co-workers,12 there is a probable involvement of PtpB in evasion of lymphocyte-mediated responses. In experiments performed with activated macrophages and guinea pigs it was observed that gene inactivation of PtpB in Mtb caused accelerated mycobacterial cell death after macrophage invasion.12
Sulfonyl derivatives are well known as antimicrobial compounds. Sulfonyl-hydrazones and sulfonamides, for example, show antibacterial activity against Staphylococcus aureus and Escherichia coli.13 In the case of sulfonamides, the action mechanism seems to be linked to inhibition of folic acid synthesis, which is essential for bacterial survival.14 The sulfonyl compounds 1, 2, 3, 4 and 5 (Fig. 1) inhibit Mtb growth by different mechanisms.2,15–20 For example, compound 1, useful for treating tuberculosis, is active by inhibiting the Mtb RNA polymerase,2 and compound 2 inhibits the branched-chain amino acid biosynthesis, due to the interaction with the enzyme Mtb acetohydroxyacid synthase.15 The sulfonamide 6 (Fig. 1, OMTS), was shown to be a selective and potent inhibitor of Mtb protein tyrosine phosphatase B (PtpB); this compound inhibits PtpB with an IC50 value of 0.44 ± 0.05 μM.21a Very recently, the group of Zhang reported that from a library of 7500 compounds, a sulfonamide was identified as a potent PtpB inhibitor.21b Also, double-site isoxazole-based compounds were described as competitive PtpB inhibitors,22 and a bidentate benzofuran salicylic acid derivative as a noncompetitive PtpB inhibitor.23
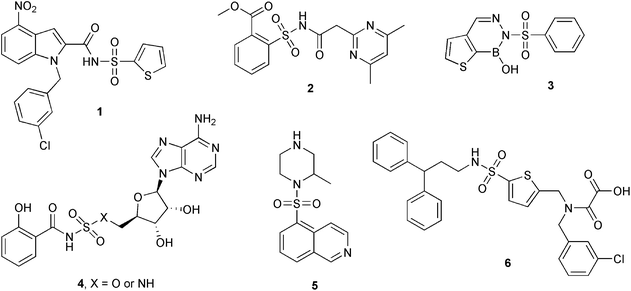 | ||
| Fig. 1 Sulfonamides active against M. tuberculosisgrowth (X = O or NH). Compound 6 (OMTS) is a selective inhibitor of MtbPtpB. | ||
In this work, six sulfonamides and eight sulfonyl-hydrazones cyclic imides derivatives were evaluated as inhibitors of Mtb protein tyrosine phosphatase B (PtpB). The sulfonamides and sulfonyl-hydrazones were obtained as shown in Fig. 2 and 3.
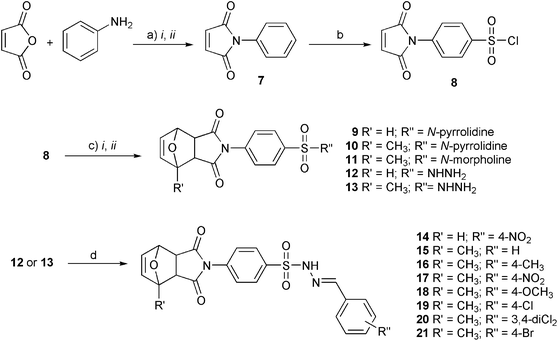 | ||
| Fig. 2 Synthesis of compounds 7–21: a) i. Et2O, r.t., 30 min.; ii. NaOAc, Ac2O, 60 °C, 1 h; b) 6 eq. HClSO3, 0–60 °C; c) i. Furan or 2-methylfuran, Et2O, r.t.; ii. MeOH, 0 °C, amines; d) benzaldehydes, EtOH, r.t., 1–3 h. | ||
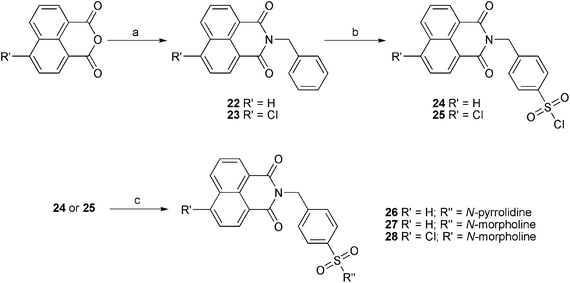 | ||
| Fig. 3 Synthesis of compounds 22–28: a) EtOH, reflux, benzylamine, 2–3 h.; b) 6 eq. HClSO3, 0–60 °C; c) MeOH, 0 °C, 2 eq. amines. | ||
For the synthesis of the sulfonamides 9, 10 and 11, and sulfonyl-hydrazones 14, 15, 16, 17, 18, 19, 20 and 21, five to six reaction stages were carried out (Fig. 2).24 The first stage consisted of the synthesis of N-phenylmaleimide (7), through the maleamic acid formation, that occurred upon maleic anhydride ring opening (Fig. 2a, i), followed by condensation with aniline, using acetic anhydride and anhydrous sodium acetate as dehydrating agents (Fig. 2a, ii). N-Phenylmaleimide (7) was then chlorosulfonated with six equivalents of chlorosulfonic acid to obtain the respective sulfonyl chloride (8). The Diels–Alder adducts were obtained by reaction between these sulfonyl chlorides and furan or 2-methylfuran. From these adducts it was possible to directly obtain the sulfonamides (9–11), with yields between 40 and 62%, as well as the sulfonyl-hydrazides (12 and 13), using two equivalents of the appropriate amine in methanol. The amines were pyrrolidine and morpholine, for the sulfonamides, and hydrazine hydrate, for the sulfonyl-hydrazides. The sulfonyl-hydrazides (12 and 13), through condensation with the appropriate benzaldehyde, formed the sulfonyl-hydrazones (14–21), with yields between 48 and 84%.
The compounds 26, 27 and 28 were synthesized by similar procedures. In the case of sulfonamides 26–28 (yields 61–74%), the cyclic imides (22 and 23) were prepared by reflux of 4-chloro-1,8-naphthalimide or 1,8-naphthalimide and benzylamine in ethanol (Fig. 3). The obtained cyclic imides were chlorosulfonated (24 and 25), followed by reaction with pyrrolidine or morpholine.
The structures of all compounds, including those still not published (21, 24, 25, 26, 27 and 28), were confirmed by chemical identification data using 1H NMR, 13C NMR, IR and elementary analysis.
The bacterial expression vector pET28-PtpB, containing the full coding sequence for PtpB, was used to produce recombinant His-tagged protein as previously described for PtpA.25,26
Inhibitor screening assays were performed using a single compound concentration (25 μM) in a continuous assay for phosphatase activity using p-nitrophenylphosphate as a substrate.25,27a,27b The in vitro inhibitory activity towards PtpB was evaluated and the results are shown in Table 1. In general, the sulfonyl-hydrazones cyclic imides derivatives (14–21) presented the best inhibitory activity towards PtpB, mainly compounds 14, 15, 19 and 21, that decrease the relative activity of the enzyme to around 40% at a compound final concentration of 25 μM. To assess the activity of the most active compounds, further assays were carried out and the IC50 values on PtpB (5–80 μM of compound) were established and are shown in Table 2. It can also be observed that the sulfonamides (9, 10, 11, 26, 27 and 28) were not able to inhibit PtpB, and the sulfonyl chloride 8 and the N-fenylmaleimide 7 presented poor inhibitory activity (Table 1).
| Compounds | PtpB Inhibition (%) |
|---|---|
| 7 | 27 ± 3 |
| 8 | 14 ± 5 |
| 9 | — |
| 10 | — |
| 11 | — |
| 14 | 57 ± 3 |
| 15 | 59 ± 2 |
| 16 | 26 ± 1 |
| 17 | 32 ± 6 |
| 18 | — |
| 19 | 59 ± 7 |
| 20 | 34 ± 2 |
| 21 | 59 ± 4 |
| 26 | — |
| 27 | — |
| 28 | — |
| Compound | IC50 (μM) | |
|---|---|---|
| PtpB | PTP1B | |
| 15 | 18 ± 1 | 7 ± 1 |
| 19 | 21 ± 2 | 41 ± 1 |
| 21 | 39 ± 1 | 88 ± 1 |
| 14 | 41 ± 1 | 245 ± 13 |
The data displayed in Table 1 indicated that the substituent in the phenyl ring affects the activity of the enzyme. Among the sulfonyl-hydrazones with a methyl group in the bicycle ring (15–21), those with electron donor substituents at phenyl ring, presented none (18) or low (16) inhibitory activity, while those without substituent or with electron acceptor substituents displayed moderate (17 and 20) or good PtpB inhibitory activity (15, 19 and 21). The data in Table 2 allowed to establish a possible relationship between the structures of these compounds and their inhibitory potency: 15 does not present substituents at the phenyl ring, and is the more active compound; 19 is the second in inhibitory capacity and has a chlorine atom at the phenyl ring, while 21 is the third in inhibitory capacity and has a phenyl ring substituted by a bromine atom. Compounds 17 and 20 have, respectively, a nitro group and two chlorine atoms at the phenyl ring, and display moderate PtpB inhibition. Based on these results, we suggest that in the sulfonyl-hydrazones with a methyl group in the bicycle ring (R′ = CH3), the inhibitory activity is lost with the increase in number and volume of the substituent at phenyl ring. Compound 14, the fourth most active, is the only one that does not have a methyl group in the bicycle ring (R′ = H), but has a nitro group at the phenyl ring as compound 17, and its action also could be impaired by the volume of nitro group.
The activity of the selected compounds towards PtpB was compared to their activity on a mammalian tyrosine phosphatase (PTP1B); compound 14 was considered selective to PtpB, 19 and 21 moderately selective, and 15 not selective (Table 2). The four most potent compounds evaluated against PtpB, 15 (IC50 = 18 μM), 19 (IC50 = 21 μM), 21 (IC50 = 39 μM) and 14 (IC50 = 41 μM), were selected for further kinetic analysis (Fig. 4).
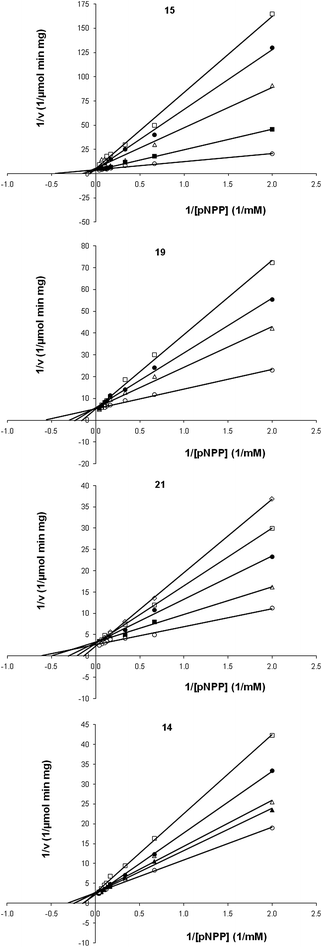 | ||
| Fig. 4 MtbPtpB experimental kinetic parameters (Vmax, Kmapp and Ki) for the substrate pNPP and compounds 14, 15, 19 and 21. Compound: 0μM (○), 5μM (▲), 10μM (■), 15μM (△), 20μM (●), 25μM (□), 30μM (◇). | ||
The mechanism of inhibition and the respective Ki values were determined. The inhibition of PtpB was found to be competitive with respect to the substrate p-nitrophenyl phosphate (pNPP), for these four compounds. The Lineweaver–Burk double-reciprocal plots (Fig. 4), obtained at three or four different inhibitor concentrations, converge at the y-axis (1/Vmax), whereas the slope (KMapp/Vmax) and x-axis intercepts (−1/KMapp) vary with inhibitor concentration. In this context, the Vmax value remains constant, and the KMapp values increase with increasing inhibitor concentrations. This behavior is consistent with a mutually exclusive binding mode, where the bioactive molecules compete with the substrate for the free enzyme active site.28 The Ki values obtained for compounds 15, 19, 21 and 14 are in the low micromolar range (7.0 μM; 2.5 μM; 9.0 μM and 15.0 μM, respectively), indicating the potential of these sulfonyl-hydrazone derivatives as new lead candidates for efficient inhibitors of PtpB.
Structure-based molecular docking simulations were performed in order to investigate the binding mode of the competitive inhibitors 15, 19, 21 and 14 into the PtpB active site, as well as to provide insights on the structural basis underlying the inhibitory potency of these compounds. The analysis of the proposed binding modes revealed that inhibitor 15, which contains a phenyl substituent, presented the best results (i.e., highest score) and was taken as a reference compound for all docking calculations. The proposed binding mode indicates that compound 15 (IC50 = 18 μM; Ki = 7.0 μM) filled the narrow channel of the catalytic Cys160, establishing extensive polar and hydrophobic contacts with the PtpB active site.21,29 Accordingly, the polar atoms of 15 formed seven hydrogen bonds to Ser57, Tyr125, Cys160, Ala162, Asp165, Arg166 (Fig. 5A). In addition, two dipole interactions assisted the stabilization of the binary complex: (i) the electronegative sulfonyl group is complementary to the helix dipole of helix α5, and (ii) an ion-dipole interaction between the hydrazone group and the side chain of Lys164 (Fig. 5A). The non-polar parts of 15 make favorable van der Waals contacts with several structural elements of the protein, some of them establishing hydrophobic interactions with the macromolecular counterparts, which significantly contributes to the complex stability. Particularly, the phenyl substituent is docked into the hydrophobic pocket formed by the amino acid residues Phe98, Leu101, Ile203, Met206, Leu227, and Val231 (Fig. 5A). A similar conformation is observed for inhibitor 19 (IC50 = 21 μM; Ki = 2.5 μM), which exhibits a p-chlorophenyl substituent favorably fulfilling the hydrophobic pocket of the PtpB (Fig. 5B). This binding mode is consistent with the crystallographic data of the OMTS inhibitor in complex with PtpB.21 In this complex, the analogous m-chlorophenyl substituent of OMTS is oriented towards the hydrophobic pocket of PtpB binding site.
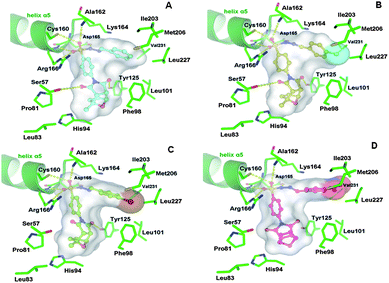 | ||
| Fig. 5 Three-dimensional models of the interactions formed by the competitive inhibitors 15 (A), 19 (B), 21 (C) and 14 (D) in the active site of MtbPtpB. The amino acid residues are depicted as stick models and the helix α5 as cartoon. The ligands are simultaneously indicated as ball-and-stick models and the Connolly surface. The yellow dashed lines indicate the hydrogen bonds between the ligands and the protein. | ||
The proposed binding mode of the compounds 21 (IC50 = 39 μM; Ki = 9.0 μM) and 14 (IC50 = 41 μM; Ki = 15.0 μM) are similar to those observed for 15 and 19. The hydrogen bonds and the dipole interactions established by the sulfonyl moiety are also observed, however, the presence of the p-bromo and p-nitrophenyl bulky substituents partially overlap with the side chain of Leu227 (Fig. 5C and 5D, respectively). Additionally, the models indicate a slightly different conformation regarding the pyrrolidine-2,5-dione rings, which is 25–35° rotated in comparison to the equivalent group of 15. In the case of 21, the rotation orients the pyrrolidine-2,5-dione group in a favorable position for a hydrogen bond, while for compound 14, the adopted conformation is unfavorable for hydrogen bonds. These molecular events may contribute to the decreased inhibitory potency of the compounds.
Taking into consideration the molecular information provided by the modeling studies, the synthesis of new inhibitors of MtbPtpB is currently underway. In particular, with the aim of fully exploring the possibility of hydrophobic interactions within the binding pocket, several derivatives with substituents at the positions ortho, meta and para of the phenyl group (e.g., CH3, F, CF3), as well as the replacement of the phenyl group with bulky groups (e.g., cyclohexyl, cycloheptyl) will be designed. A similar synthetic approach has been described previously yielding PtpB inhibitors with improved inhibitory activity.30 The molecular modeling studies indicated that the new inhibitors candidates showed similar binding modes, filling the hydrophobic pocket of the protein by the establishment of van der Waals contacts, thereby giving a significant contribution to the complex stability (data not shown).
In conclusion, synthetic compounds were obtained and assayed towards PtpB. Four sulfonyl-hydrazones N-phenylmaleimide derivatives displayed excellent activity (compounds 15, 19, 21 and 14), and the inhibition of PtpB was found to be competitive with respect to the substrate p-nitrophenyl phosphate, with Ki values in low micromolar range. Structure-based molecular docking simulations were performed and indicated that the new inhibitors showed similar binding modes, filling the hydrophobic pocket of the protein by the establishment of van der Waals contacts, thereby giving a significant contribution to the complex stability. The sulfonyl-hydrazones N-phenylmaleimide derivatives described here appear as a new class of lead compounds potentially interesting as novel candidates for the treatment of TB.
Acknowledgements
The authors are grateful to CNPq, CAPES, MCT, FINEP and FAPESC for financial support and fellowships. This research was supported by grants and fellowships from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), CAPES (Coordenação de Pessoal de Nível Superior), FAPESC (Fundação de Apoio à Pesquisa Científica do Estado de Santa Catarina), MCT (Ministério da Ciência e Tecnologia) e FINEP (Financiadora de Estudos e Projetos). We would like to thank the Department of Chemistry from Universidade Federal de Santa Catarina for some facilities.References
- D. Maher and M. Raviglione, Clin. Chest Med., 2005, 26, 167–182 CrossRef.
- Y. L. Janin, Bioorg. Med. Chem., 2007, 15, 2479–2513 CrossRef CAS.
- T. R. Frieden, T. R. Sterling, S. S. Munsiff, C. J. Watt and C. Dye, Lancet, 2003, 362, 887–899 CrossRef.
- J. M. Grange and A. R. Zumla, J. R. Soc. Promot. Health, 2002, 122, 78–81 Search PubMed.
- L. Ballell, R. A. Field, K. Duncan and R. J. Young, Antimicrob. Agents Chemother., 2005, 49, 2153–2163 CrossRef.
- K. Duncan and C. E. Barry, Curr. Opin. Microbiol., 2004, 7, 460–465 CrossRef CAS.
- A. E. Greenstein, C. Grundner, N. Echols, L. M. Gay, T. N. Lombana, C. A. Miecskowski, K. E. Pullen, P. Y. Sung and T. Alber, J. Mol. Microbiol. Biotechnol., 2005, 9, 167–181 CrossRef CAS.
- V. V. Vintonyak, A. P. Antonchick, D. Rauh and H. Waldmann, Curr. Opin. Chem. Biol., 2009, 13, 272–283 CrossRef CAS.
- L. Tabernero, A. R. Aricescu, E. Y. Jones and S. E. Szedlacsek, FEBS J., 2008, 275, 867–882 CrossRef CAS.
- I. DeVinney, I. Steele-Mortimer and B. B. Finlay, Trends Microbiol., 2000, 8, 29–33 CrossRef CAS.
- Z. Y. Zhang, Curr. Opin. Chem. Biol., 2001, 5, 416–423 CrossRef CAS.
- R. Singh, V. Rao, H. Shakila, R. Gupta, A. Khera, N. Dhar, A. Singh, A. Koul, Y. Singh, M. Naseema, P. R. Narayanan, C. N. Paramasivan, V. D. Ramanathan and A. K. Tyagi, Mol. Microbiol., 2003, 50, 751–762 CrossRef CAS.
- D. J. Bhatt, G. C. Kamdar and A. R. Parikh, J. Indian Chem. Soc., 1984, 2, 788–795.
- R. J. Henry, Bacteriol. Rev., 1943, 7, 175–262 CAS.
- J. A. Grandoni, P. T. Marta and J. V. Schloss, J. Antimicrob. Chemother., 1998, 42, 475–482 CrossRef CAS.
- C. W. Levy, C. Baldock, A. J. Wallace, S. Sedelnikova, R. C. Viner, J. M. Clough, A. R. Stuitje, A. R. Slabas, D. W. Rice and J. B. Rafferty, J. Mol. Biol., 2001, 309, 171–180 CrossRef CAS.
- J. J. De Voss, K. Rutter, B. G. Schroeder, H. Su, Y. Zhu and C. E. Barry, Proc. Natl. Acad. Sci. USA, 2009, 7, 1252–1257.
- J. A. Ferreras, J. S. Ryu, F. Di Lello, D. S. Tan and L. E. Quadri, Nat. Chem. Biol., 2005, 1, 29–32 CrossRef CAS.
- R. V. Somu, H. Boshoff, C. Qiao, E. M. Bennett, C. E. Barry and C. C. Aldrich, J. Med. Chem., 2006, 49, 31–34 CrossRef CAS.
- S. J. Drews, F. Hung and Y. Av-Gay, FEMS Microbiol. Lett., 2001, 205, 369–374 CrossRef CAS.
- (a) C. Grundner, D. Perrin, R. H. Huijsduijnen, D. Swinnen, J. Gonzalez, C. L. Gee, T. N. Wells and T. Alber, Structure, 2007, 15, 499–509 CrossRef CAS; (b) L. Chen, B. Zhou, S. Zhang, L. Wu, Y. Wang, S. G. Franzblau and Z.-Y. Zhang, ACS Med. Chem. Lett., 2010, 1, 355–359 Search PubMed.
- N. J. Beresford, D. Mulhearn, B. Szczepankiewicz, G. Liu, M. E. Johnson, A. Fordham-Skelton, C. Abad-Zapatero, J. S. Cavet and L. Tabernero, J. Antimicrob. Chemother., 2009, 63, 928–36 CrossRef CAS.
- B. Zhou, Y. He, X. Zhang, J. Xu, Y. Luo, Y. Wang, S. G. Franzblau, Z. Yang, R. J. Chan, Y. Liu, J. Zheng and Z. Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4573–4578 CrossRef CAS.
- K. N. Oliveira and R. J. Nunes, Synth. Commun., 2006, 36, 3401–3409 CrossRef.
- L. D. Chiaradia, A. Mascarello, M. Purificação, J. Vernal, M. N. S. Cordeiro, M. E. Zenteno, A. Villarino, R. J. Nunes, R. A. Yunes and H. Terenzi, Bioorg. Med. Chem. Lett., 2008, 18, 6227–6230 CrossRef CAS.
- A. Mascarello, L. D. Chiaradia, J. Vernal, A. Villarino, R. V. C. Guido, P. Perizzolo, V. Poirier, D. Wong, P. G. A. Martins, R. J. Nunes, R. A. Yunes, A. D. Andricopulo, Y. Av-Gay and H. Terenzi, Bioorg. Med. Chem., 2010, 18, 3783–3789 CrossRef CAS.
- (a) M. Manger, M. Scheck, H. Prinz, J. P. von Kries, T. Langer, K. Saxena, H. Schwalbe, A. Fuerstner, J. Rademann and H. Waldmann, Chem. Bio Chem., 2005, 6, 1749–1753 CrossRef CAS; (b) D. F. McCain and Z. Y. Zhang, Methods Enzymol., 2002, 345, 507–518.
- R. A. Copeland, Evaluation of enzyme inhibitors in drug discovery, Wiley Interscience, New Jersey, 2005, pp 48–81 Search PubMed.
- C. Grundner, H. L. Ng and T. Alber, Structure, 2005, 13, 1625–1634 CrossRef CAS.
- M. B. Soellner, K. A. Rawls, C. Grundner, T. Alber and J. Ellman, J. Am. Chem. Soc., 2007, 129, 9613–9615 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental protocols. See DOI: 10.1039/c0md00253d/ |
| This journal is © The Royal Society of Chemistry 2011 |