trans-Stilbenoids: potent and selective inhibitors for human cytochrome P450 1B1†
Young-Jin
Chun
a,
Chaemin
Lim
b,
Seul Ong
Ohk
a,
Ji Min
Lee
b,
Jin Hee
Lee
c,
Sun
Choi
c and
Sanghee
Kim
*b
aCollege of Pharmacy, Chungang University, 221 Huksuk, Seoul, 156-756, Korea
bCollege of Pharmacy, Seoul National University, San 56-1, Shilim, Kwanak, Seoul, 151-742, Korea. E-mail: pennkim@snu.ac.kr; Fax: +82 2 888 0649; Tel: +82 2 880 2487
cCollege of Pharmacy, Division of Life & Pharmaceutical Sciences, and National Core Research Center for Cell Signaling & Drug Discovery Research, Ewha Womans University, 11-1 Daehyun, Seodaemun, Seoul, 120-750, Korea
First published on 8th March 2011
Abstract
On the basis of our previous insights into the structural requirements of stilbenoids for the inhibition of cytochrome P450 1B1 (CYP1B1), a series of 2,4-dimethoxy group-containing stilbenes was prepared and evaluated for their inhibitory effects on the activity of CYP1s with the ultimate goal of identifying a potent and selective CYP1B1 inhibitor. Among the thirteen derivatives prepared, five compounds exhibited similar or greater potency compared to the previous lead compound, 2,4,3′,5′-tetramethoxystilbene (2,4,3′,5′-TMS), in inhibiting CYP1B1. In particular, 2,2′,3′,4,6′-pentamethoxystilbene was found to be a more selective and more potent CYP1B1 inhibitor than 2,4,3′,5′-TMS. 2,4,2′,6′-TMS showed remarkably potent inhibitory activity against CYP1B1 (IC50 = 1.77 ± 0.14 nM) and also had a very high selectivity toward CYP1 isoenzymes. Molecular modeling was performed to determine the key molecular interactions with the CYP1B1 and CYP1A2 structures. On the basis of these structural and biological studies, the design of more potent and more selective drug-like derivatives can be envisaged.
Introduction
The cytochrome P450 subfamily 1 (CYP1) has long been of interest to researchers because of its relevance to carcinogenesis. The human CYP1 family contains three members, CYP1A1, CYP1A2, and CYP1B1. Among these, CYP1B1 has received considerable attention as a new drug target.1–6CYP1B1 is a major enzyme for the 4-hydroxylation of 17β-estradiol (E2) to produce a carcinogenic metabolite, 4-hydroxyestradiol (4-OHE2).1–8CYP1B1 is also involved in the metabolic activation of various endogenous compounds and of a variety of polycyclic aromatic hydrocarbons (PAHs) and heterocyclic aromatic amines.1–6,9–12Several studies have shown that CYP1B1 is expressed at a higher level in malignant tumor cells in the kidney, breast, brain, and lungs than in normal cells.13–16 Overexpression of CYP1B1 has also been detected in colorectal adenomas and premalignant prostate tumors. Moreover, a recent study revealed that CYP1B1 up-regulation plays an important role in endometrial carcinogenesis by targeting multiple pathways.17 Because CYP1B1 is up-regulated in premalignant and malignant tumors, CYP1B1 might be involved in the early development of cancer. In addition, CYP1B1 could be involved in anticancer drug resistance because the metabolism of various anticancer agents by CYP1B1 causes the suppression of the pharmacological effects of drugs.18 Because CYP1B1 is thought to be more detrimental than beneficial,19 the availability of potent and selective inhibitors of CYP1B1 would facilitate the development of CYP1B1-targeted cancer chemotherapy and prevention drugs.
Owing to the important role of CYP1B1 in chemical carcinogenesis, various studies have attempted to identify potent and selective inhibitors from natural sources in recent years.1,20–23 For instance, the inhibitory effects of natural stilbenoids on CYP1 activity have been studied.3,24 It was reported that resveratrol 1 (Fig. 1) inhibited CYP1A1 and CYP1B125–27 and that rhapontigenin 2 exhibited a strong and selective inhibition of CYP1A1 over CYP1B1.3Pinostilbene 3, desoxyrhapontigenin 4, and pterostilbene 5 also appear to be inhibitors of CYP1B1, with Ki values of 0.9, 2.1, and 0.9 μM, respectively, although these compounds showed more potent inhibition of CYP1A1 (Ki = 0.12, 0.16, and 0.57 μM, respectively).28
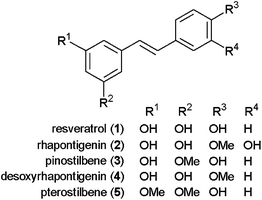 | ||
| Fig. 1 Structures of natural stilbenoids that inhibit CYP1s. | ||
On the basis of studies of natural stilbenoids showing that the selectivities and inhibitory potencies of the compounds were sensitive to the substitution patterns on the trans-stilbene template, we have previously prepared and evaluated a series of lipophilic stilbenoid compounds of general structure 6 (Fig. 2) in which the phenyl ring at site A contains dimethoxy groups at the 3′ and 5′ positions.29 This study allowed us to identify a highly potent and selective competitive inhibitor of CYP1B1 with an IC50 value of 6 nM: 2,4,3′,5′-tetramethoxystilbene 7 (2,4,3′,5′-TMS).30 2,4,3′,5′-TMS also showed suppression of CYP1B1 induction by TCDD in MCF-7 cells and HL-60 cells.31 In addition, our previous studies revealed interesting structure–activity relationships (SAR) in terms of the importance of substituents at the ortho position of the phenyl ring at site B for discriminating between CYP1As and CYP1B1.
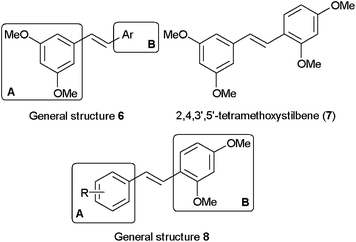 | ||
| Fig. 2 General structures 6 and 8 and the structure of 2,4,3′,5′-tetramethoxystilbene (7). | ||
Our prior SAR study led us to analyze stilbenoid compounds with general structure 8 (Fig. 2), in which the phenyl ring at site B contains dimethoxy groups at the 2 and 4 positions, as a new lead template for further chemical optimization and structure–activity studies. As part of our ongoing program to develop potent and selective CYP1B1 inhibitors, we now describe the biological evaluation of a series of 2,4-dimethoxy group containing stilbenes.
Results and discussion
Chemistry
The designed compounds were prepared in high overall yield (90–97%) using our previously reported column chromatography-free solution phase synthetic pathway (Scheme 1).29 Briefly, Horner–Wadsworth–Emmons reactions between phosphonate 9 and various aromatic aldehydes 10 (1.1 equiv) yielded a mixture of Z and Eolefin 11. After a standard aqueous work-up, the excess aldehyde 10 was removed from the mixture by treatment with Girard's reagent T 12. The trans-stilbene derivative 8 was obtained from the Z/E mixture of 11 by heating with catalytic amounts of iodine in refluxing heptane. The NMR spectra of the synthesized trans-stilbenes showed that the products were very clean, and the HPLC analysis indicated that the products were pure enough for biological testing (>97%).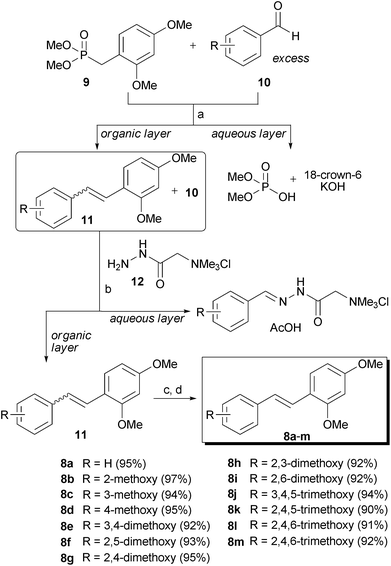 | ||
| Scheme 1 Synthesis of trans-stilbene derivatives 8a–m: Reagents and conditions: (a) 18-crown-6, KOH, CH2Cl2, rt; (b) 12, AcOH, CH2Cl2, rt; (c) cat. I2, heptane, reflux; (d) NaHSO3, aqueous work-up. | ||
In vitro assays
To examine the effects of 2,4-dimethoxy group-containing stilbenes on the activities of human cytochrome P450 1A1, 1A2, and 1B1, the changes in ethoxyresorufinO-deethylation (EROD) activities were measured using the E. colimembranes expressed human recombinant P450 1A1, 1A2, or 1B1 with human NADPH-P450 reductase.32,33 As indicated in Table 1, although the inhibitory potency and selectivity varied based on the substitution pattern, all of the compounds preferentially inhibited CYP1B1 over CYP1A1 and CYP1A2. These results validated our assumption that the presence of a 2-methoxy group may play a very important role in selective binding to the active site of CYP1 isozymes.| Compdb | R | IC50/nMc | Ratio (1A1/1B1) | Ratio (1A2/1B1) | ||
|---|---|---|---|---|---|---|
| 1A1 | 1A2 | 1B1 | ||||
| a Enzyme activities were measured as described in the ESI.† b Compounds 7, 8a, 8d, 8e, 8g, 8i, 8j, and 8k were previously known. c The IC50 values are the means ± range of two separate experiments determined using a quadratic expression of nonlinear regression methods using Graph-Pad Prism software (San Diego, CA). Control activities in the absence of chemical were 20, 5, and 2 nmol of resorufin formed min−1 (nmol of P450)−1 for CYP1A1, CYP1A2, and CYP1B1, respectively. d Taken from ref. 29. | ||||||
| 7 d | See Fig. 2 | 300 ± 20 | 3100 ± 880 | 6 ± 2 | 50 | 517 |
| 8a | H | 493 ± 104 | 326 ± 64 | 25.0 ± 1.0 | 20 | 13 |
| 8b | 2-Methoxy | 336 ± 61 | 792 ± 136 | 15.5 ± 1.6 | 22 | 51 |
| 8c | 3-Methoxy | 98.2 ± 24.4 | 903 ± 71 | 15.2 ± 1.5 | 6.5 | 59 |
| 8d | 4-Methoxy | 666 ± 242 | 1060 ± 239 | 57.5 ± 25.1 | 12 | 18 |
| 8e | 3,4-Dimethoxy | 190 ± 76 | 337 ± 30 | 41.8 ± 7.9 | 4.6 | 8.1 |
| 8f | 2,5-Dimethoxy | 350 ± 83 | 302 ± 28 | 3.51 ± 0.40 | 100 | 86 |
| 8g | 2,4-Dimethoxy | 222 ± 22 | 204 ± 23 | 8.81 ± 1.52 | 25 | 23 |
| 8h | 2,3-Dimethoxy | 192 ± 21 | 247 ± 36 | 60.3 ± 11.0 | 3.2 | 4.1 |
| 8i | 2,6-Dimethoxy | 350 ± 49 | 170 ± 32 | 1.77 ± 0.14 | 194 | 95 |
| 8j | 3,4,5-Trimethoxy | 301 ± 83 | 4690 ± 220 | 71.0 ± 5.0 | 4.2 | 66 |
| 8k | 2,4,5-Trimethoxy | 78.4 ± 8.8 | 236 ± 18 | 5.46 ± 2.03 | 14 | 43 |
| 8l | 2,4,6-Trimethoxy | 148 ± 14 | 188 ± 35 | 4.93 ± 1.50 | 30 | 38 |
| 8m | 2,3,6-Trimethoxy | 420 ± 104 | 1740 ± 220 | 3.30 ± 0.70 | 127 | 526 |
Several compounds exhibited similar or improved potencies compared to the previous lead compound 7 in inhibiting CYP1B1. Compound 8m, 2,2′,3′,4,6′-pentamethoxystilbene, was found to be a more selective and more potent CYP1B1 inhibitor than 7. It showed a potent inhibitory effect on CYP1B1 (IC50 = 3.30 ± 0.70 nM) and, to a much lesser extent, on CYP1A1 (IC50 = 420 ± 104 nM) and CYP1A2 (IC50 = 1740 ± 220 nM). The differences between CYP1A1, CYP1A2, and CYP1B1 inhibition are statistically significant as determined by ANOVA (p < 0.05). Compound 8i, 2,4,2′,6′-TMS, showed a remarkably potent inhibitory activity against CYP1B1 (IC50 = 1.77 ± 0.14 nM) and had a very high selectivity toward the CYP1 isozymes. It exhibited the greatest selectivity (194-fold) of 1B1 over 1A1, but the selectivity between 1B1 and 1A2 was diminished compared to that of 8m (95-fold vs. 526-fold).
The mono-methoxylated phenyl ring-containing compounds, 8b–8d, as well as the non-substituted phenyl ring-containing compound 8a, also preferentially inhibited 1B1 over 1A1 and 1A2. However, the inhibitory potentials and selectivities were diminished substantially compared to those of 8m and 8i. Among the mono-methoxylated compounds, 3-methoxyphenyl ring-containing compound 8c had the lowest selectivity for CYP1B1 over CYP1A1, and 4-methoxylated compound 8d had the lowest selectivity for CYP1B1 over CYP1A2. These results suggest that the substituent at the 3 or 4 position might have a negative effect on the selectivity for CYP1B1 inhibition. This detrimental effect of the methoxy group at the 3 or 4 position was further evidenced by the 3,4-dimethoxylated compound 8e, which exhibited a very low selectivity towards the CYP1 isoenzymes.
Among the dimethoxylated compounds, compound 8h, having dimethoxy groups at the 2 and 3 position, was the least active against CYP1B1 and exhibited the lowest selectivity. However, compound 8i, which differed structurally from 8h by only the position of one methoxy group (C-3vs.C-6) showed remarkably greater inhibitory activity and selectivity. These observations demonstrated that the exact locations of methoxy groups are a very important feature for selectivity.
The tri-methoxylated compounds generally showed potent inhibition of CYP1B1; 8j was the exception. The selectivities varied and were largely dependent on the position of the methoxy groups. Of the tri-methoxylated compounds examined, compound 8m exhibited the greatest selectivity and potency.
Computational study
To understand why one of the most active compounds, 8m, is over 500-fold more selective for CYP1B1 compared with CYP1A2, we performed a docking study using the CYP1B134 and CYP1A235 structures. As shown in Fig. 3a, 8m occupied the active site of CYP1B1 very well. The binding pocket near the heme, including Ala133 and Val395, appears to be large enough to accommodate the trimethoxy phenyl group of 8m. There are two hydrogen bonds between 8m and the residues of CYP1B1; the meta-methoxy group was able to form a hydrogen bond with Thr510, and the para-methoxy group in the other phenyl ring could make a hydrogen bond with Thr325, which is located in the upper side of the binding pocket.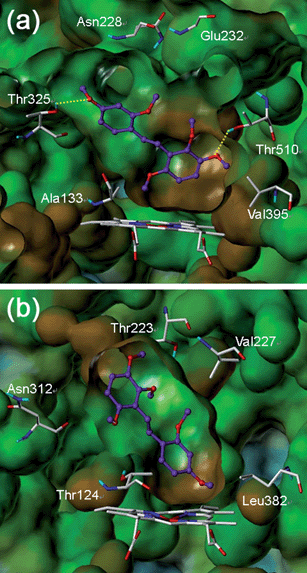 | ||
| Fig. 3 Predicted binding modes of 8m in (a) CYP1B1 and (b) CYP1A2. The key interacting residues are marked and displayed in capped sticks with carbon atoms in white. The ligand 8m is depicted as a ball-and-stick structure with carbon atoms in purple. Hydrogen bonds are shown by yellow dashed lines. The Van der Waals surfaces of the ligand were generated by MOLCAD and are colored based on hydrogen bonding properties (blue: H-bond-accepting regions). The Fast Connolly surfaces of the proteins were generated by MOLCAD and are colored based on lipophilic potential properties, ranging from brown (highest lipophilic area) to blue (highest hydrophilic area). Protein surfaces are Z-clipped, and non-polar hydrogens are undisplayed for clarity. | ||
In contrast, the docking result of 8m in CYP1A2 revealed a flipped binding mode (Fig. 3b). In CYP1A2, the binding pocket near the heme is narrower than that of CYP1B1. This narrowness might be why the bulky trimethoxy phenyl ring of 8m was docked in the opposite direction, thus avoiding the steric repulsion that would otherwise be encountered at the binding pocket. Although compound 8m could occupy the active site of CYP1A2, there is no hydrogen bonding between 8m and the residues of CYP1A2. This lack of a vital hydrogen bonding might be the reason for the weak inhibitory activity of 8m against CYP1A2. Our docking model successfully illustrated the selectivity of 8m for CYP1 inhibition and offered important information for developing new stilbenoid compounds targeting CYP1B1.
Conclusion
In conclusion, a series of stilbenoids was prepared and evaluated as to their effects on the activity of CYP1s with the ultimate goal of identifying a potent and selective CYP1B1 inhibitor. Among the compounds tested, 2,2′,3′,4,6′-pentamethoxystilbene (8m) was found to be the most selective and potent CYP1B1 inhibitor to date. Compound 8i also showed a remarkably potent inhibitory activity against CYP1B1 and had a very high selectivity toward the CYP1 isozymes. These compounds exhibited improved selectivity and potency profiles compared to the previous lead compound 2,4,3′,5′-TMS 7, which made them excellent tools to characterize the enzymatic properties of CYP1B1 and to evaluate the effects of CYP1B1 inactivation on various diseases. Our results of structure–activity relationship and docking studies are of interest for allowing the design of drug-like stilbene derivatives that have a superior selectivity and activity. Further studies are currently in progress, including a more detailed biological evaluation and chemical optimizations.Acknowledgements
We would like to thank Prof. D. F. Lewis for providing the coordinates of the CYP1B1 model. This work was supported by Basic Science Research program (No. 20100027312) and NCRC program (R15-2006-020) through the National Research Foundation of Korea (NRF) funded by the Korea government (MEST).Notes and references
- Y.-J. Chun and S. Kim, Med. Res. Rev., 2003, 23, 657 CrossRef CAS.
- R. D. Bruno and V. C. Njar, Bioorg. Med. Chem., 2007, 15, 5047 CrossRef CAS.
- F. P. Guengerich, Y.-J. Chun, D. Kim, E. M. J. Gillam and T. Shimada, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2003, 523–524, 173 CrossRef.
- T. M. Sissung, D. K. Price, A. Sparreboom and W. D. Figg, Mol. Cancer Res., 2006, 4, 135 CrossRef CAS.
- J. A. Barnett, D. L. Urbauer, G. I. Murray, G. N. Fuller and A. B. Heimberger, Clin. Cancer Res., 2007, 13, 3559 CrossRef CAS.
- M. C. E. McFadyen, W. T. Melvin and G. I. Murray, Mol. Cancer Ther., 2004, 3, 363 CAS.
- C. L. Hayes, D. C. Spink, B. C. Spink, J. Q. Cao, N. J. Walker and T. R. Sutter, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9776 CrossRef CAS.
- T. Shimada, J. Watanabe, K. Kawajiri, T. R. Sutter, F. P. Guengerich, E. M. Gillam and K. Inoue, Carcinogenesis, 1999, 20, 1607 CrossRef CAS.
- V. Vasiliou and F. J. Gonzalez, Annu. Rev. Pharmacol., 2008, 48, 333 Search PubMed.
- F. F. Parl, S. Dawling, N. Roodi and P. S. Crooke, Ann. N. Y. Acad. Sci., 2009, 1155, 68 CrossRef CAS.
- M. C. McFadyen and G. I. Murray, Future Oncol., 2005, 1, 259 Search PubMed.
- T. Shimada and Y. Fujii-Kuriyama, Cancer Sci., 2004, 95, 1 CrossRef CAS.
- G. I. Murray, M. C. Taylor, M. C. McFadyen, J. A. McKay, W. F. Greenlee, M. D. Burke and W. T. Melvin, Cancer Res., 1997, 57, 3026 CAS.
- J. A. McKay, W. T. Melvin, A. K. Ah-See, S. W. Ewen, W. F. Greenlee, C. B. Marcus, M. D. Burke and G. I. Murray, FEBS Lett., 1995, 374, 270 CrossRef CAS.
- M. C. E. McFadyen, M. E. Cruickshank, I. D. Miller, H. L. McLeod, W. T. Melvin, N. E. Haites, D. Parkin and G. I. Murray, Br. J. Cancer, 2001, 85, 242 CrossRef CAS.
- T. Tokizane, H. Shiina, M. Igawa, H. Enokida, S. Urakami, T. Kawakami, T. Ogishima, S. T. Okino, L.-C. Li, Y. Tanaka, N. Nonomura, A. Okuyama and R. Dahiya, Clin. Cancer Res., 2005, 11, 5793 CrossRef CAS.
- S. Saini, H. Hirata, S. Majid and R. Dahiya, Cancer Res., 2009, 69, 7038 CrossRef CAS.
- T. Shimada, M. Iwasaki, M. V. Martin and F. P. Guengerich, Cancer Res., 1989, 49, 3218 CAS.
- D. W. Nebert and T. P. Dalton, Nat. Rev. Cancer, 2006, 6, 947 CrossRef CAS.
- Y. J. Moon, X. Wang and M. E. Morris, Toxicol. in Vitro, 2006, 20, 187 CrossRef CAS.
- T. K. H. Chang, J. Chen and E. Y. Yeung, Toxicol. Appl. Pharmacol., 2006, 213, 18 CrossRef CAS.
- H. Y. Chan, Z.-Y. Chen, D. S. C. Tsang and L. K. Leung, Biomed. Pharmacother., 2002, 56, 269 CrossRef CAS.
- H. Takemura, T. Itoh, K. Yamamoto, H. Sakakibara and K. Shimoi, Bioorg. Med. Chem., 2010, 18, 6310 CrossRef CAS.
- B. B. Aggarwal, A. Bhardwaj, R. S. Aggarwal, N. P. Seeram, S. Shishodia and Y. Takada, Anticancer Res., 2004, 24, 2783 CAS.
- Y. J. Chun, M. Y. Kim and F. P. Guengerich, Biochem. Biophys. Res. Commun., 1999, 262, 20 CrossRef CAS.
- T. K. H. Chang, W. B. K. Lee and H. H. Ko, Can. J. Physiol. Pharmacol., 2000, 78, 874 CrossRef CAS.
- S. R. Beedanagari, I. Bebenek, P. Bui and O. Hankinson, Toxicol. Sci., 2009, 110, 61 CrossRef CAS.
- R. Mikstacka, D. Przybylska, A. M. Rimando and W. Baer-Dubowska, Mol. Nutr. Food Res., 2007, 51, 517 CrossRef CAS.
- S. Kim, H. Ko, J. E. Park, S. Jung, S. K. Lee and Y.-J. Chun, J. Med. Chem., 2002, 45, 160 CrossRef CAS.
- Y.-J. Chun, S. Kim, D. Kim, S.-K. Lee and F. P. Guengerich, Cancer Res., 2001, 61, 8164 CAS.
- Y.-J. Chun, S.-K. Lee and M. Y. Kim, Drug Metab. Dispos., 2005, 33, 1771 CAS.
- J. T. M. Buters, S. Sakai, T. Richter, T. Pineau, D. L. Alexander, U. Savas, J. Doehmer, J. M. Ward, C. R. Jefcoate and F. J. Gonzalez, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1977 CrossRef CAS.
- J. T. M. Buters, B. Mahadevan, L. Quintanilla-Martinez, F. J. Gonzalez, H. Greim, W. M. Baird and A. Luch, Chem. Res. Toxicol., 2002, 15, 1127 CrossRef CAS.
- D. F. V. Lewis, E. M. J. Gillam, S. A. Everett and T. Shimada, Chem.-Biol. Interact., 2003, 145, 281 CrossRef CAS.
- S. Sansen, J. K. Yano, R. L. Reynald, G. A. Schoch, K. J. Griffin, C. D. Stout and E. F. Johnson, J. Biol. Chem., 2007, 282, 14348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods and full characterization for all new compounds. 1H and 13C NMR spectra for selected compounds. See DOI: 10.1039/c0md00242a/ |
| This journal is © The Royal Society of Chemistry 2011 |