Interaction mechanisms of CdTe quantum dots with proteins possessing different isoelectric points†
Zhisong
Lu
ab,
Weihua
Hu
ab,
Haifeng
Bao
ab,
Yan
Qiao
ab and
Chang Ming
Li
*ab
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Singapore, 637457, Singapore. E-mail: Ecmli@ntu.edu.sg; Fax: + 65 6791 1761; Tel: + 65 6790 4485
bCentre for Advanced Bionanosystems, Nanyang Technological University, 70 Nanyang Drive, Singapore, 637457, Singapore
First published on 14th February 2011
Abstract
Interactions of surface-immobilized and solution-dispersed quantum dots (QDs) with proteins possessing different isoelectric points, particularly the molecular binding capability and agglomeration of proteins with QDs are examined, indicating an electrostatic attraction nature. A mechanism involving protein surface charge distribution and protein molecular size is elucidated.
Quantum dots (QDs) are semiconductor crystalline clusters with a size range of 1 nm to 10 nm.1 In the past decade, QDs have been widely used in cell imaging,2 advanced biosensing,3 and single molecule detection4 because of their superior optical properties over conventional organic fluorescent dyes. Recently, there has been a growing interest of utilizing QDs for in vivo applications such as in vivo animal tracking,5 tumor targeting6 and photosensitizing nanodrugs for cancer photodynamic therapy.7 In those applications, nanocrystals circulate around the body along with blood, finally locating at target organs. Blood acts as both matrix and carrier of the injected QDs. Since various proteins always exist in blood, protein adsorption on QD surface is unavoidable, possibly making QDs losing specificity, reducing fluorescent efficiency and even causing severe immune responses. Therefore, understanding of interaction of QDs with proteins and its mechanism could provide medicinal chemistry to design optimal QD nanodrugs for biomedical applications.
Interactions between CdSe/ZnS QD and human serum albumin (HSA),8CdSe/ZnS QD and bovine serum albumin (BSA),9 CdS QD and BSA,10 CdS QD and haemoglobin (Hb),11CdTe QD and HSA12 and CdSe QD and Hb13 have been reported. However, those studies only focus on the interaction of QDs with different proteins, but do not relate the interactions to the specific properties of proteins. The mechanism, which can be utilized to predict the binding of different proteins with QDs, is still obscure.
In this study, mercaptosuccinic acid (MSA)-capped CdTe QDs (Fig. S1†)14 and three blood proteins [glucose oxidase (GOD), haemoglobin (Hb) and cytochrome c (Cyt C)] with different isoelectric points (IEPs) are selected to investigate the interactions using surface plasmon resonance (SPR), atomic force microscopy (AFM), photoluminescence (PL) spectrometry, and hydrodynamic size/zeta potential measurements. Based on the experimental results and protein structures, a mechanism for protein-QD interaction is elucidated.
QD-attached gold and platinum surface have been widely applied for in vivo biosensors.15 The protein binding on the QD-immobilized surface is sensed by an in situSPR method, in which QDs are immobilized on a SPR gold disk through covalent binding with amino groups of a self-assembled monolayer of cysteamine on the gold disk surface. The amino groups are activated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride for the covalent bonding of QDs, and the surface is incubated with deionized water for 30 min to remove intermediates and to regenerate carboxyl groups on QD surface for remaining experiments.
In situ SPR-measured binding curves (Fig. 1) are used to demonstrate real-time adsorption profiles of proteins. The SPR angle increases very fast during the binding process of Hb, whereas only slight SPR angle enhancements for adsorptions of both Cyt C and GOD are observed, indicating distinct adsorption capabilities of different proteins on the QD-modified gold disk. The SPR angle changes can be converted to surface mass changes and absorbed protein density changes as shown in ESI Table S1.† An alteration of 100 millidegrees corresponds to a surface concentration of about 1 ng mm−2 for most proteins.16 Molecular weights of GOD, Hb and Cyt C are 160.0, 64.5 and 12.4 kDa, respectively. The surface protein molecular densities of GOD, Hb and Cyt C of ∼3.2 × 109, ∼7.0 × 1010 and ∼1.0 × 1011 molecules mm−2 were calculated by equation S1 in the ESI.†SPR data demonstrate that the three proteins can interact with QDs, and their binding capability is Cyt C > Hb > GOD. The modified surface morphology was examined using AFM, illustrating well-distributed immobilized small QDs with a very high surface density (Fig. 2A). Fig. 2B–D are surface morphological images of GOD, Hb and Cyt C adsorbed disks, respectively, of which the big islands should be the proteins. It indicates that the molecular binding capability to adsorb proteins onto QD-immobilized surface is Cyt C > Hb > GOD, and the surface coverage of Cyt C is the highest, which is consistent to the SPR measurement. It is worthy of a note that the gold surface is covalently immobilized by QDs. A new electric double layer is eventually formed on the most outside QD surface for the interaction with proteins.
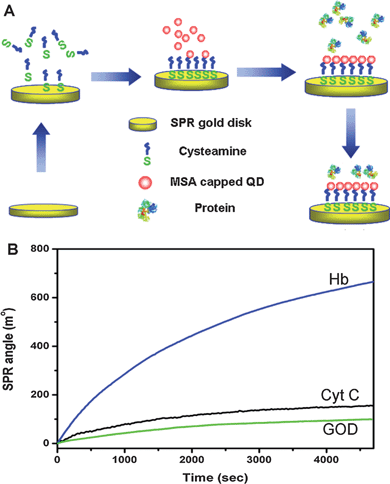 | ||
| Fig. 1 (A) The schematic diagram of cysteamine self-assembly, QDs covalent immobilization and protein adsorption. (B) Real-time SPR binding curves of adsorptions of GOD, Hb and Cyt C on a QD immobilized SPR gold disk. | ||
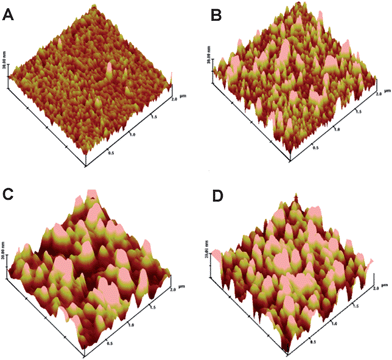 | ||
| Fig. 2 Morphologies of QDs immobilized SPR gold disk (A), QDs-SPR gold disk with GOD adsorption (B), QDs-SPR gold disk with Hb adsorption (C), QDs-SPR gold disk with Cyt C adsorption (D). Z scale = 30 nm. | ||
The adsorption process of proteins on surface-immobilized QDs may not be the same as that on spherical shape QDs in blood or body fluid. Hence, interactions of solution-dispersed QDs with proteins were investigated. It has been reported by us that peptide or amino acid adsorption on the surface of QDs can quench their fluorescences.17 Thus, the binding effects of proteins with QDs in solution was probed by measuring fluorescence emission from QDs. Mixing of QDs with different concentrations of GOD does not change PL spectra (ESI Fig. S2†). It is probably due to the weak interaction between QDs and GOD. However, a strong concentration-dependent fluorescence quenching occurs in both QD-Hb and QD-Cyt C mixtures (Fig. 3), suggesting that the QD-protein interactions are much stronger than QD-GOD ones. Stern–Volmer equation is used to simulate the fluorescence quenching effect:18
| I0/I = 1 + Ksv [Q] = 1 + kq τ0 [Q] | (1) |
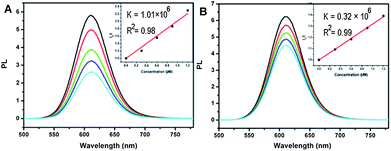 | ||
| Fig. 3 (A) PL spectra of QDs – Hb mixtures with different protein concentrations. (B) PL spectra of QDs – Cyt C mixtures with different protein concentrations. Insets correspond to Stern – Volmer plots. | ||
SPR and AFM results show that Cyt C has the highest molecular binding amount on the surface-attached QDs. However, Hb is found to have the highest fluorescence quenching capability in PL spectra assay. The hydrodynamic sizes of QDs, proteins and QD-protein combinants were examined to further understand the fluorescence quenching effect. Fig. 4 shows that the average hydrodynamic sizes of QDs, GOD, Hb and Cyt C are 6.00, 10.17, 8.46 and 4.66 nm, respectively. The QD–GOD combinant through bindings has an average size of 9.05 nm, which is between the sizes of QDs and GOD. Curves in Fig. 4A illustrate that the size distribution of the QD–protein combinants covers both size ranges of QDs and GOD, indicating that the binding of QDs and GOD is very weak. In this case most of the GOD molecules still remain their original sizes, and thus the QD-GOD curve covers the size distribution of GOD. The broader peak and the enhanced volume percentage in a relatively small size range attribute to the free QDs in the QD-protein mixture solution. The average size for the QD-Cyt C combinant is 9.27 nm, denoting the binding of QDs and Cyt C and the formation of QD-Cyt C complex (Fig. 4C). These results are greatly in agreement with the discoveries in SPR and AFM measurements from both surface-attached and solution-dispersed QDs. Interestingly, the complex of QD and Hb displays a size distribution peak above 1 μm, which is over 100 times larger than that of QD or Hb (Fig. 4B), suggesting the agglomeration of the protein molecules and QD nanocrystals in the solution. It can be speculated that QDs in the mixture solution may function as linkers to connect Hb molecules to form microsized clusters, thus resulting in the significant quenching effect. The substrate-attached QDs can be used to measure the protein binding capabilities, but the agglomeration is not able to be observed due to the lack of free QDs as linkers, further supporting that the large sized aggregates of QDs and proteins in solutions is resulted from QD-linking. The fluorescence quenching and hydrodynamic size measurements can examine the interactions of QDs and proteins in solutions, but they cannot determine different QD-protein binding capability. Thus, it is necessary to use both substrate-immobilized and solution-dispersed QDs in studies to better understand interactions of QDs with proteins.
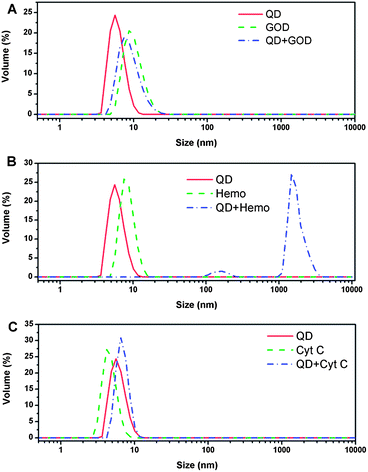 | ||
| Fig. 4 (A) Hydrodynamic size distributions of QDs, GOD and QDs-GOD complex; (B) Hydrodynamic size distributions of QDs, Hb (Hemo) and QDs-Hb complex; (C) Hydrodynamic size distributions of QDs, Cyt C and QDs-Cyt C complex. | ||
No observed shift of amide I (1658 cm−1) and amide II (1550 cm−1) absorption peaks in FTIR spectra (ESI Fig. S3†) indicates that QDs and proteins do not form hydrogen bonds. The electrostatic attraction may be the binding force for interactions of GOD, Hb and Cyt C with QDs. To further verify the role of electrostatic force, effects of solution pH on QD-protein interactions were investigated. The pH values are selected according to the IEPs of GOD (IEP = 4.2), Hb (IEP = 6.8) and Cyt C (IEP = 10.0). SPR binding curves in Fig. 5A–C demonstrates that solution pH can change the adsorption rate and amount of the proteins. For each protein, the lower the pH is, the higher the binding capability is. Zeta potentials of proteins and QDs in phosphate buffer with different pHs represent that QDs are negatively charged at all pHs, but proteins possess more positive charges at pH 4.3 (Fig. 5D). This agrees well with the pH dependent SPR result. Results from both SPR and zeta potential measurements at different pHs show that proteins can interact with QDs through the electrostatic attraction. The amino acid side chains and the structure of the protein may also influence the interaction due to stereo-hindrance effects, which are under investigation in our lab.
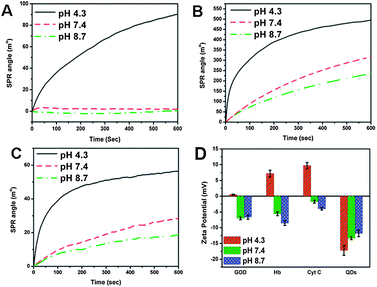 | ||
| Fig. 5 The pH dependent interactions of QDs with GOD (A), Hb (B) and Cyt C (C); Zeta potentials of QD, GOD, Hb and Cyt C in phosphate buffer with different pH (D). | ||
The IEP of a protein is determined by amino acids of the peptide chains, which have distinct charges and may locate at the inner or outer part of the protein molecule since the protein exists in solution with a 3-dimensional conformation. Thus, some proteins with a lower IEP may still possess significant amount of positive charges on their outer surfaces at a neutral pH. Fig. 6 illustrates the space topologies of GOD, Hb and Cyt C at pH 7.4. The dominant charges on the outer surface of GOD (IEP = 4.2) and Cyt C (IEP = 10.0) are negative and positive, respectively. Thus, the interaction capability of GOD with QDs is the lowest, while that of Cyt C is the highest among three proteins. However, although the IEP of Hb is about 6.8, there is still significant amount of positive charge on the molecule surface, which may result in its strong interaction with the negatively charged QDs. Interestingly, the solution-based agglomeration discussed above only occurs in the QD-Hb interaction. Even if Cyt C possesses positive charges as that of Hb does, the agglomeration is not observed in the QD-Cyt C interaction. This is very possibly caused by the larger size of Hb (Fig. 6), which is able to provide more sites for QD binding. Therefore, the surface positive charge distribution may mainly contribute to the protein interaction with QDs. In a brief, the QD-protein interactions may rely not only on the IEP of a protein, but also on the surface charge distributions and the protein sizes. Since most of protein sequences and structures are available freely from the online database, the analysis of surface charge distribution and the molecular size using bioinformatics tools may benefit the prediction of QD-protein interactions to design high performance of QD-protein systems in biomedical applications.
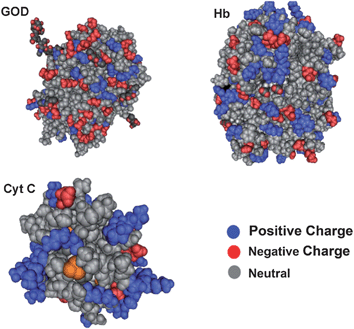 | ||
| Fig. 6 Structural representations of GOD (A), Hb (B) and Cyt C (C). | ||
In summary, interactions of QDs with proteins are measured to indicate an electrostatic attraction nature. The experimental results show that both surface- and solution-based assays need to be carried out for better understanding of QD-protein interactions including the binding capabilities and the possibility of aggregate formation. For the first time a mechanism involving protein surface charge distribution and protein molecular size is elucidated to explain the QD-protein interaction through electrostatic attraction. The analysis of protein sequences and structures using bioinformatics tools may benefit the prediction of QD-protein interactions. This work provides fundamental insights for interactions of nanoscaled QDs with biological systems, an important part of medicinal chemistry, which could be used in design of QD-based nanodrugs. Because various nanoparticles have demonstrated an increasing importance in biological, biomedical and pharmaceutical applications,21 the mechanism may also be extended to explain and predict the interaction of proteins with other nanoparticles if electrostatic attraction is the binding force. QD binding may change the protein structures, subsequently affecting their functions. Thus, the QD-induced protein structure alteration and the QD loading capacity of proteins should be investigated in future studies.
Acknowledgements
The authors thank Center for Advanced Bionanosystems, Nanyang Technological University, Singapore for the financial support.Notes and references
- (a) C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS; (b) A. P. Alivisatos, Science, 1996, 271, 933 CrossRef CAS; (c) A. P. Alivisatos, J. Phys. Chem., 1996, 100, 13226 CrossRef CAS; (d) A. L. Efros, M. Rosen, M. Kuno, M. Nirmal, D. J. Norris and M. Bawendi, Phys. Rev. B: Condens. Matter, 1996, 54, 4843 CrossRef CAS; (e) M. Nirmal, D. J. Norris, M. Kuno, M. G. Bawendi, A. L. Efros and M. Rosen, Phys. Rev. Lett., 1995, 75, 3728 CrossRef CAS; (f) H. F. Bao, X. Q. Cui, C. M. Li and J. F. Zang, Nanotechnology, 2007, 18, 455701 CrossRef; (g) R. Hardman, Environ. Health Perspect., 2006, 114, 165 CrossRef; (h) R. Li, C. M. Li, H. F. Bao and Q. L. Bao, Appl. Phys. Lett., 2007, 91, 223901 CrossRef.
- V. Biju, D. Muraleedharan, K. I. Nakayama, Y. Shinohara, T. Itoh, Y. Baba and M. Ishikawa, Langmuir, 2007, 23, 10254 CrossRef CAS.
- C. A. Constantine, K. M. Gattas-Asfura, S. V. Mello, G. Crespo, V. Rastogi, T. C. Cheng, J. J. DeFrank and R. M. Leblanc, Langmuir, 2003, 19, 9863 CrossRef CAS.
- S. M. Stavis, J. B. Edel, K. T. Samiee and H. G. Craighead, Lab Chip, 2005, 5, 337 RSC.
- X. Gao, Y. Cui, R. M. Levenson, L. W. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS.
- (a) K. C. Weng, C. O. Noble, B. Papahadjopoulos-Sternberg, F. F. Chen, D. C. Drummond, D. B. Kirpotin, D. Wang, Y. K. Hom, B. Hann and J. W. Park, Nano Lett., 2008, 8, 2851 CrossRef CAS; (b) E. B. Voura, J. K. Jaiswal, H. Mattoussi and S. M. Simon, Nat. Med., 2004, 10, 993 CrossRef CAS.
- (a) R. Bakalova, H. Ohba, Z. Zhelev, M. Ishikawa and Y. Baba, Nat. Biotechnol., 2004, 22, 1360 CrossRef CAS; (b) R. Bakalova, H. Ohba, Z. Zhelev, T. Nagase, R. Jose, M. Ishikawa and Y. Baba, Nano Lett., 2004, 4, 1567 CrossRef CAS.
- Q. Xiao, S. Huang, Z. D. Qi, B. Zhou, Z. K. He and Y. Liu, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1020 CrossRef CAS.
- M. M. Dzagli, V. Canpean, M. Iosin, M. A. Mohou and S. Astilean, J. Photochem. Photobiol., A, 2010, 215, 118 CrossRef CAS.
- M. Amelia, R. Flamini and L. Latterini, Langmuir, 2010, 26, 10129 CrossRef CAS.
- X. C. Shen, X. Y. Liou, L. P. Ye, H. Liang and Z. Y. Wang, J. Colloid Interface Sci., 2007, 311, 400 CrossRef CAS.
- J. B. Xiao, Y. L. Bai, Y. F. Wang, J. W. Chen and X. L. Wei, Spectrochim. Acta, Part A, 2010, 76, 93 CrossRef.
- D. H. Hu, H. M. Wu, J. G. Liang and H. Y. Han, Spectrochim. Acta, Part A, 2008, 69, 830 CrossRef.
- (a) Z. S. Lu, C. M. Li, H. F. Bao, Y. Qiao, Y. H. Toh and X. Yang, Langmuir, 2008, 24, 5445 CrossRef CAS; (b) H. F. Bao, Z. S. Lu, X. Q. Cui, Y. Qiao, J. Guo, J. M. Anderson and C. M. Li, Acta Biomater., 2010, 6, 3534 CrossRef CAS; (c) X. D. Cao, C. M. Li, H. F. Bao, Q. L. Bao and H. Dong, Chem. Mater., 2007, 19, 3773 CrossRef CAS; (d) Z. S. Lu, Y. Qiao, X. T. Zheng, M. B. Chan-Park and C. M. Li, Med. Chem. Commun., 2010, 1, 84 RSC; (e) H. F. Bao, E. K. Wang and S. J. Dong, Small, 2006, 2, 476 CrossRef CAS.
- (a) C. G. Shi, J. J. Xu and H. Y. Chen, J. Electroanal. Chem., 2007, 610, 186 CrossRef CAS; (b) L. H. Tang, Y. H. Zhu, X. L. Yang, J. J. Sun and C. Z. Li, Biosens. Bioelectron., 2008, 24, 319 CrossRef CAS.
- (a) Z. S. Lu, C. M. Li, Q. Zhou, Q. L. Bao and X. Q. Cui, J. Colloid Interface Sci., 2007, 314, 80 CrossRef CAS; (b) S. Suire, M. C. Maurel and F. Guillou, Eur. J. Biochem., 1996, 239, 52 CrossRef CAS.
- Z. S. Lu, C. M. Li, H. F. Bao, Y. Qiao and Q. L. Bao, J. Nanosci. Nanotechnol., 2009, 9, 3252 CrossRef CAS.
- S. J. Clarke, C. A. Hollmann, F. A. Aldaye and J. L. Nadeau, Bioconjugate Chem., 2008, 19, 562 CrossRef CAS.
- Y. Zhang, L. Mi, P. N. Wang, J. Ma and J. Y. Chen, J. Lumin., 2008, 128, 1948 CrossRef CAS.
- Y. P. Wang, Y. L. Wei and C. Dong, J. Photochem. Photobiol., A, 2006, 177, 6 CrossRef CAS.
- (a) X. Q. Cui, C. M. Li, H. F. Bao, X. T. Zheng and Z. S. Lu, J. Colloid Interface Sci., 2008, 327, 459 CrossRef CAS; (b) K. Y. Wong and X. L. Liu, Med. Chem. Commun., 2010, 1, 125 RSC; (c) K. Matsui, M. Karasaki, M. Segawa, S. Y. Hwang, T. Tanaka, C. Ogino and A. Kondo, Med. Chem. Commum., 2010, 1, 209 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, physical properties of synthesized MSA capped QDs, PL spectra of QDs-GOD mixture, Fourier transform infrared spectra, surface density of each protein on a QDs immobilized SPR gold disk (Table S1). See DOI: 10.1039/c0md00237b |
| This journal is © The Royal Society of Chemistry 2011 |